Interleukin-23 and interleukin-17: Importance in pathogenesis and therapy of psoriasis
Published Web Location
https://doi.org/10.5070/D33n39n8xmMain Content
Interleukin-23 and interleukin-17: Importance in pathogenesis and therapy of psoriasis
Parvathi Mudigonda1 BS, Tejaswi Mudigonda1 BS, Ashley N Feneran1 DO, Habibollah S Alamdari1 BA, Laura Sandoval1 DO, Steven R Feldman1,2,3 MD PhD
Dermatology Online Journal 18 (10): 1
1. Department of Dermatology2. Department of Pathology
3. Department of Public Health Sciences
Center for Dermatology Research, Wake Forest School of Medicine, Winston-Salem, North Carolina
Abstract
Emerging data in mice and humans reveals a critical contribution of Th17-associated cytokines, particularly interleukin-(IL)-23 and IL-17, in the pathogenesis of psoriasis. The IL-23/Th17 pathway is a therapeutic target for biologic agents and systemic therapies in psoriasis treatment. A literature search was performed to review and summarize the current evidence on IL-17 and IL-23 as a basis for understanding the use of anti-IL-17 and anti-IL-23 agents for psoriasis therapy. Using PubMed, recent articles were identified pertaining to IL-17, IL-23, and psoriasis. Signaling via the heterodimeric IL-23 receptor induces production of IL-17, which stimulates production of proinflammatory keratinocyte cytokines that mediate the psoriatic response. An overexpression of IL-23, IL-17, or Th17 cells in transgenic mice is associated with the development of inflammatory disease. Both IL-17 knockout mice and humans with a genetic IL-17 deficiency are susceptible to extracellular and intracellular pathogens. This suggests a potential for adverse effects from clinical administration of anti IL-23-p40/IL-17 therapies. Anti-p40 antibodies, briakinumab and ustekinumab, were tolerated in clinical trials and substantially improved psoriasis. Further trials of anti IL-17 therapies are needed to assess their clinical use and potential for infection and other adverse events.
Introduction
Psoriasis is a chronic, immunologically mediated disease with a multifactorial genetic basis [1, 2, 3]. This inflammatory and hyperproliferative disease of the skin and joints results from the infiltration of activated dendritic cells (DC) and T cells and the ensuing release of inflammatory cytokines by these and other cells, including keratinocytes (KCs) and endothelial cells [4, 5, 6, 7]. Until recently, interferon-γ (IFN-γ)-producing T helper (Th) 1 cells were considered to be the main pathogenic cells in psoriasis. However, newer studies have revealed that IL-23 stimulates and promotes differentiation in a subset of memory-activated CD4+ T-cells known as Th17 cells [8, 9, 10, 11]. This IL-23/Th17 pathway plays an important role in the pathogenesis of psoriasis (Figure 1).
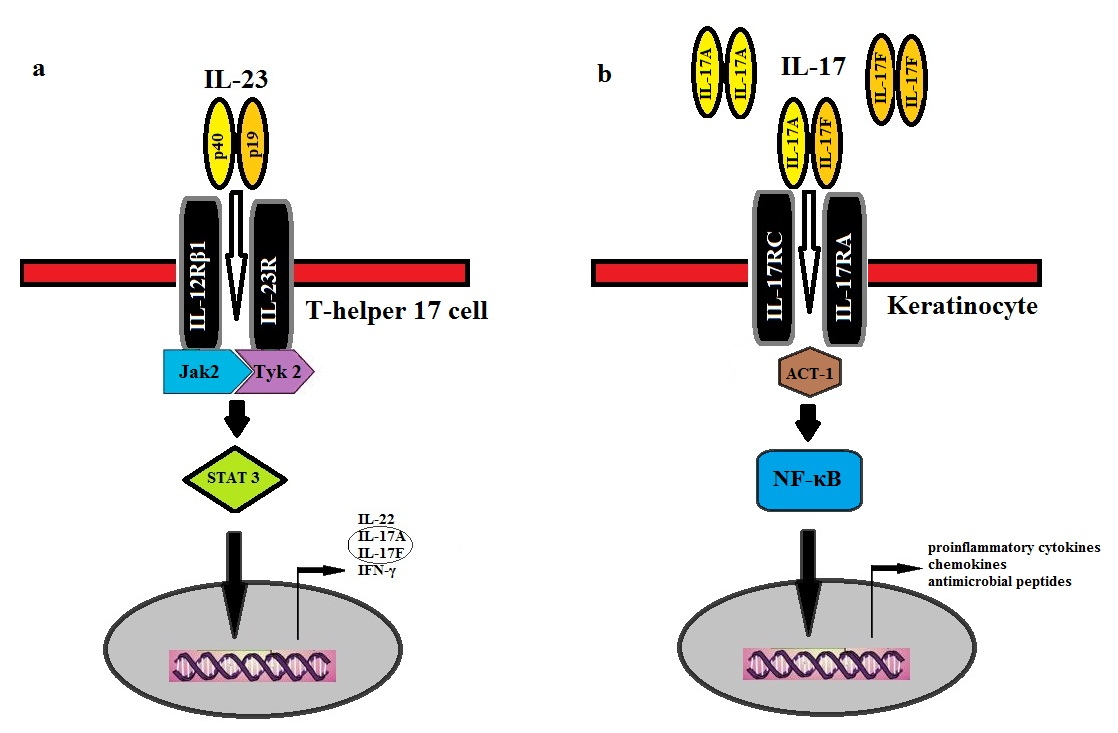 | 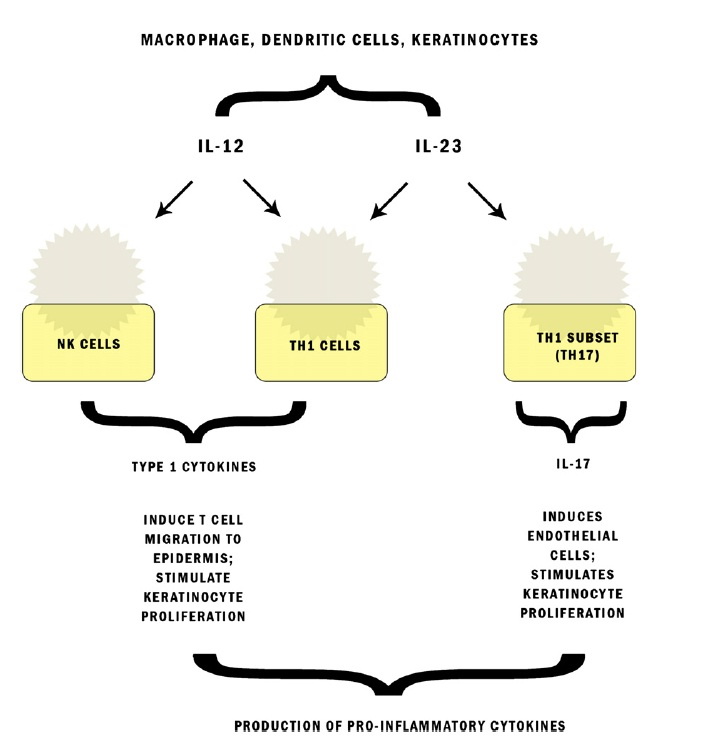 |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. Pathways engaged by interleukin (IL)-12, IL-23, and IL-17. NK, Natural killer; TH1- T-helper 1. Adapted with permission
from: Torti DC, Feldman SR. Interleukin-12, interleukin-23, and psoriasis: current prospects. J Am Acad Dermatol. Dec 2007;57(6):1059-1068. Figure 2. Signal transduction pathways engaged by interleukin (IL)-23 and IL-17 cytokines. (a) Interleukin (IL)-23, a heterodimeric cytokine composed of p40 and p19 subunits, binds to its IL-23 receptor located on T-helper (Th) 17 cells. The IL-23 receptor complex is composed of IL-23R and IL-12Rβ1 subunits, which are associated with the Jak family members, Jak 2 and Tyk2, respectively. Intracellular phosphorylation signaling activates STAT3 molecules, which translocate into the nucleus and upregulate the transcription of cytokines of IL-17A, IL-17F, IL-22, and IFN-γ. (b) Binding of IL-17A and IL-17F either as homodimers or heterodimers, to the IL-17R complex, composed of IL-17RC and IL-17RA, induces the recruitment of the adapter protein, Act1. Subsequent intracellular phosphorylation activity activates the transcription factor, NF-κB, which enters the nucleus and directs the transcription of proinflammatory cytokines, chemokines, and antimicrobial peptides. | |
IL-23 is a heterodimeric protein comprised of two subunits, IL-23p19 and IL-12p40. IL-23 drives the Th17 response by binding and signaling through its receptor complex composed of the IL-12Rβ1 and IL-23R subunits (Figure 2). IL-12 and IL-23 share the IL-12p40 subunit, encoded by the IL12B gene, and the IL-12 and IL-23 receptors share the common IL-12Rβ1 subunit. IL-23R is mainly expressed by T cells, natural killer cells, and to a lesser extent by monocytes and DCs [12]. When the IL-23 receptor (IL-23R), located on memory-T cells, is activated, it promotes the development of Th17 cells, characterized by the production of IL-17A, IL-17F, and IL-22 [5, 13, 14]. Signal transduction in Th17 cells occur through the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling molecules. IL-17A and IL-17F signal through the heterodimeric IL-17R (IL-17 receptor) complex on keratinocytes as either homodimers or heterodimers. Activated IL-17R, composed of IL-17RC and IL-17RA, induces the recruitment of the adapter protein, Act1. Subsequent production of numerous proinflammatory cytokines, chemokines, and antimicrobial peptides drive the inflammatory response in psoriatic lesions [15]. The purpose of this study was to review the literature on IL-23 and IL-17 as a basis for understanding the use of anti-IL-23 and anti-IL-17 therapies for psoriasis. Using PubMed, relevant articles were selected from January 2000 to October 2011 elucidating information on the IL-23/Th17 pathway through either murine or human models. These articles describe immunological pathways and genetic influences, as well as discuss how current and future therapies address the role of IL-23 and IL-17 in psoriatic disease.
IL-23/Th17 cells in the pathogenesis of psoriasis: Evidence from mouse models
Murine models have helped elucidate the role of IL-17 and IL-23 in psoriasis-like disease (Table 1). Different approaches have been used in order to induce the development of psoriasis-like lesions in mice. These include the adoptive transfer of human CD4+CD45Rbhigh T cells into immunodeficient scid/scid recipient mice [16], injection of recombinant murine (rm) IL-17F [17] or IL-23 [18-22] into murine ear tissue, the application of 12-O-tetradeconylphorbol-12-acetate (TPA) to K5.Stat3C [23] or K14/VEGF [24] transgenic mice, the topical application of imiquimod (IMQ) [25], and xenotransplantation of grafts from psoriasis patients onto mice [26].
Immunohistochemisty and real-time reverse transcriptase polymerase chain reaction profiling of the cytokines in the IL-23/Th17 pathway have provided evidence for the pathogenesis of psoriasis. Transcript levels of IL-17A, IL-22, IL-17F, IL-23p19, and IL-12/IL-23p40 were elevated in mice with psoriasis-like lesions induced by TPA, IMQ, or rmIL-23 when compared to the levels of these cytokines in wild-type mice and were comparable to those found in human psoriatic lesions [19, 23, 24, 27]. Examination of the dermis of TPA-induced lesions revealed increased Th17 (CD4+IL-17+) cells, whereas the rmIL-23-induced lesions showed increased IL-17-producing CD3+ T cells [19-23]. IL-22, in particular, is a key cytokine produced by Th17 cells. Previous in vitro studies demonstrate that IL-22 acts synergistically with IL-17 to enhance KC release of neutrophil-recruiting chemokines and expression of antimicrobial peptides [28, 29, 30]. Furthermore, the administration of IL-17F into mouse skin also increased the expression of another cytokine, IL-8, which is upregulated in human psoriatic lesions [17].
The role of the IL-23/Th17 pathway in psoriasis is further demonstrated by the use of targeted antibodies and mice with receptor deficiencies. IMQ-induced psoriatic inflammation was completely blocked in mice deficient for the IL-23 or the IL-17 receptor [27]. Mice deficient in the IL-17 receptor developed reduced TPA-induced psoriatic lesions [23]. In addition, the administration of anti-IL-17A antibodies markedly inhibited the development of IL-23 or TPA induced psoriasiform inflammation or epidermal hyperplasia [19, 23].
The xenotransplant model may be the best model of psoriasis, in a sense humanizing mice by using transplanted human skin [31]. Immunosuppressed mice with asymptomatic grafts from humans with psoriasis develop typical features of psoriasis over several weeks [31, 32]. A recent study found an increase in IL-23 and IL-23R on CD3+ T cells that coincided with the development of psoriasiform lesions in xenotransplanted mice. Furthermore, human IL-23-specific monoclonal antibodies inhibited these lesions from developing and showed an efficacy comparable to that of anti-TNF blockers [26].
Although there are no definitive animal models of psoriasis, collectively, these disease models suggest that the Th17 pathway (IL-23, IL-23R, IL-17, IL-17R) can be targeted to treat psoriasis.
IL-17 and IL-23 in the pathogenesis of psoriasis: Evidence from human psoriatic lesions
Studies of human psoriatic lesions also find a role for IL-17 and IL-23 in psoriasis (Table 1). IL-17 production by CD4+ T cells is induced by IL-23 secreted by dendritic cells in the dermis of patients with psoriasis (Figure 1) [30, 33]. Analysis of psoriatic lesions by immunohistochemistry demonstrated high levels of IL-23p19 protein in cells with DC morphology distributed throughout the dermal layers [30]. An additional study isolated dermal single cell suspensions of normal skin (n=3) and psoriasis lesions (n=3) in order to demonstrate the effects of dermal DCs on inducing intracellular Th17 cytokine production in allogenic CD4+ T cells [34]. After culturing dermal DC with allogenic CD4+ T cells, psoriatic DCs induced a far greater number of CD4+ T cells to produce IL-17 compared with normal dermal DCs [34]. Flow cytometric analysis has also been used to compare circulating primary CD4+ cells isolated from healthy individuals to those in individuals with untreated psoriasis [35]. IL-17A + IFN-γ−, IL-17A + IL-22 + IFN-γ−, and IL-17A + IL-22−IFN-γ− cells were increased in psoriasis, indicative of elevations in circulating Th17 cells.
Numerous studies comparing lesional and nonlesional biopsies from psoriasis patients have reported elevated mRNA expression for IL-17A, IL-17F, IL-22, and IFN-γ in lesional skin [30, 36, 37, 38]. Enzyme-linked immunosorbent assay (ELISA) serum analysis comparing 122 psoriasis patients and 78 healthy controls revealed significantly higher levels of tumor necrosis factor (TNF)-α, IFN-γ, IL-2, IL-6, IL-7, IL-8, IL-12, IL-17, IL-18, and vascular endothelial growth factor (VEGF) in psoriasis patients [39]. Furthermore, levels of TNF-α, IFN-γ, IL-12, IL-17, IL-18, and VEGF correlated with the severity of psoriasis as determined by the Psoriasis Area Severity Index (PASI) score. These cytokines subsequently decreased after psoriasis treatments, which included topical corticosteroid, topical vitamin D3, psoralen plus ultraviolet A, etretinate, and cyclosporine [39].
Although IL-17 producing CD4+ T cells serve as a major participant in psoriasis pathogenesis, IL-17 producing CD8+ T (Tc17) cells may also play an important role. Tc17 cells are significantly higher in psoriasis plaques than in skin from healthy individuals. Similar to Th17 cells, epidermal CD8+ IL-17+ cells from psoriasis-inflamed skin produce Th17-related cytokines (IL-17, IL-21, and IL-22) [40, 41]. In addition, these CD8+ IL-17+ T cells appear to have some resistance to suppression from T regulatory (Treg) cells, proliferating much faster than CD8+ IL-17− T cells [40]. Overall, this selective increase of Tc17 cells in psoriatic skin, particularly in the epidermis, reinforces previous studies’ claims of the importance of CD8+ T cells in the pathogenesis of psoriasis [42, 43].
Higher levels of effector molecules induced by IL-17 are also linked to psoriasis. A gene array profile analysis showed IL-17 induced the upregulation of the genes for antimicrobial peptides, cytokines, and neutrophil-attracting CXCL chemokines in keratinocytes [44]. IL-6 is one of the proinflammatory cytokines upregulated by the IL-17F-IL-17R interaction in KCs [45]. Elevated IL-6 mRNA is thought to play a role in psoriasis development by rendering effector T cells refractive to suppression by Treg cells [45, 46, 47]. Another important effector molecule is CC chemokine ligand (CCL20). This CC chemokine receptor (CCR)6 ligand is believed to be a key stimulus for attracting CCR6+ immature DCs and CCR6+ Th17 cells from blood into inflamed cutaneous tissue [48, 49, 50]. In a human keratinocyte monolayer model, Th17 cytokines (IL-17A, IL-22, or TNF-α) stimulated the upregulation of CCL20 [51]. Other chemokines, such as CXCL1, CXCL3, CXCL5, CXCL6, and CXCL8 were also found to be significantly induced in keratincoytes by IL-17 treatment [52]. Taken together, these results possibly explain the continual persistence of Th17 cells in psoriasis through a Treg suppression model and a continual positive chemotactic loop mediated by chemokines.
Genetic support for the role of IL-17 and IL-23 in psoriasis
Early work on genetic polymorphic markers in Th17-associated cytokines and receptors provide new insight for the genetic basis of psoriasis (Table 1). Genetic changes that would be expected to induce IL-23, IL-23R, IL-17, or IL-17R might help one day in selecting targeted therapy based on an individual’s genetic makeup. In the case of IL-23, genetic haplotypes with specific single nucleotide polymorphisms (SNPs) that encode for the IL-23 receptor and the shared IL-12p40 subunit of IL-12 and IL-23 were identified. Individuals homozygous for these missense SNPs were found to be at increased risk of psoriasis [53, 54, 55, 56]. Polymorphisms of the IL-23R are implicated with other autoimmune diseases such as inflammatory bowel disease, rheumatoid arthritis, Graves ophthalmopathy, and ankylosing spondylitis [57-61].
In the case of IL-17, only two reports have investigated the genetic link between IL-17 pathway molecules and psoriasis [62, 63]. In a cohort of 153 unrelated Japanese psoriasis vulgaris patients, the association of IL-17 SNP with psoriasis vulgaris was assessed. A SNP in the IL-17F gene, which is located on 6p12, did not influence disease susceptibility in the Japanese population. However, provided the small sample size and lack of any other studies directly evaluating IL-17 SNPs with psoriasis, further studies are needed to investigate larger populations and other isoforms of Il-17 (IL-17A-IL-17E). A genetic link has been shown with effector molecules in the Th17 pathway through a genome-wide association analysis of SNPs for patients with psoriasis vulgaris (472 cases and 1,146 controls) and psoriatic arthritis (1,922 cases and 8,037 controls). An intrionic SNP located in the TRAF3IP2 gene locus on 6q21 was associated with both variants of psoriasis [63]. The TRAF3IP2 gene encodes the adaptor protein, ACT1, which is involved in the signal transduction pathway induced by IL-17 in keratinocytes (Figure 2).
Insights from IL-17/IL-17R/IL-23 deficiency states in murine models
Whereas blocking IL-17, the IL-17 receptor, and IL-23 may improve psoriasis, the safety of doing so must be properly assessed. Current treatment with biologics carries warnings of serious infections including tuberculosis and infections caused by bacteria, viruses, or fungi. Knockout mice may provide important pharmacologic insight into the potential adverse effects that could result from the clinical administration of anti-IL-17/IL-17R/IL-23 treatments. Mice deficient in IL-17/IL-17R/IL-23 have heightened susceptibility to infection (Table 2).
IL-17/IL-17R/IL-23 deficiency and bacterial infections in mice
IL-17 plays an immunoprotective role against intracellular Mycobacterium bovis Bacille Calmette-Guerin (BCG) infection. IL-17A gene knock-out mice failed to develop mature granulomas in mycobacterium infected lung, a process that is essential to sequestration and killing if the infection [64]. These IL-17 deficient mice also showed an impaired protective response to virulent Mycobacterium tuberculosis. Signaling via Toll-like receptors (TLR), particularly TLR2 has an impact on the increased regulation of IL-23p19 expression in response to M. tuberculosis [65]. Diminished Th17 cell responses to M. tuberculosis infection were seen in TLR2 deficient mice, as well as a reduced number of IL-17 induced cytokines that participate in recall response to the bacteria [65]. These results suggest that IL-17 serves as an important cytokine in the induction of optimal Th responses against mycobacterial infection.
Studies elucidate that IL-17 stimulates another cytokine, IL-8, which is responsible for the chemotaxis of neutrophils and the subsequent gastric inflammatory response [66, 67]. In comparing wild-type mice with IL-17 −/− knockout mice, the degree of neutrophil infiltration and the number of H. pylori colonized in the stomach of wild-type mice was significantly higher than that of the IL-17 −/− knockout mice [68]. This suggests that IL-17 may play an important role in the inflammatory response to H. pylori infection, a major cause of chronic gastritis and peptic ulcer disease.
Whereas knockout gene models provide genetic support for the role of the IL-17 pathway against bacterial infection, the use of viruses provides insight into how an underlying viral infection changes host immune responses to bacterial infections. In one study, knockout mice deficient in the IL-17 receptor were observed to have an impaired clearance of Staphylococcus aureus bacteria compared with wild-type mice, suggesting a critical role for Th17 cells against S. aureus pneumonia [69]. In the same experiment, wild type mice co-infected with influenza A and S. aureus had decreased IL-17, IL-22, and IL-23 levels, suggesting a novel mechanism by which influenza A-induced type I IFNs inhibit Th17 immunity [69]. Similarly, rhesus macques co-infected with Simian Immunodeficiency Virus (SIV) and Salmonella typhimurium bacteria showed depletion of the Th17 response compared with SIV-negative macques, which showed high expression of IL-17 after S. typhimurium infection [70]. Overall, these results further support the importance of the Th17 immune response against bacterial pathogens.
IL-17/IL-17R/IL-23 deficiency and parasitic infections in mice
Toxoplasma gondii is an opportunistic parasite that triggers an immune response characterized by neutrophil recruitment to the site of infection. The issue of whether IL-17 or signaling through IL-17R provides a protective role against T. gondii infections is debated. In one study, IL 17R-deficient mice recruit a suboptimal level of neutrophils to the site of infection and are less effective at clearing T. gondii parasite load compared to wild-type mice [71]. In contrast, another study found that IL-17 antibody neutralization enhanced survival in mice by attenuating severe inflammatory damage and early death [72]. In spite of these discrepant findings, the studies agree that IL-17/IL-17R deficiency reduces T. gondii-associated inflammation in the ileum and other major organ sites [71, 72].
IL-17 is also linked to cutaneous Leishmaniasis major and Trypanosoma cruzi infections. In IL-17-deficient mice infected with L. major, lesions were significantly smaller compared with those of wild-type mice [73]. However, IL-17-deficient mice infected with T. cruzi developed multiple organ failures, lower expression of cytokines, worse parasitemia, and a much lower survival rate compared with wild-type mice [74]. Based on these results, it appears that that the role of IL-17 is parasite-specific in terms of providing either a protective or detrimental role in the inflammatory immune response.
IL-17/IL-17R/IL-23 deficiency and viral infections in mice
IL-17/IL-17R immunity is also linked to pathogenicity of several viruses in animal models, including herpes simplex virus-1 (HSV-1) and simian AIDS [75, 76]. One study observed that IL-17R deficient mice infected with high-dose HSV-1 virus had a transient decrease in the expression of pro-inflammatory mediators, neutrophil migration, and corneal pathology [75]. However, control of virus growth in the cornea was not compromised; the clearance efficacy of HSV-1 virus 4 days post-infection was similar in both wild-type and IL-17R mice [75].
IL-17/IL-17R/IL-23 deficiency and fungal infections in mice
The role of the IL-23/Th17 pathway was implicated in fungal infections in experiments using IL-23p19−/− knockout mice models. IL-23-deficient mice infected with Pneumocystis jiroveci displayed transient but impaired clearance of infection and also a decrease of lymphocyte cytokines in their lungs compared with wild-type mice. Additionally, the administration of either anti-IL-23p19 or anti-IL-17 neutralizing antibody to wild-type mice infected with P. jiroveci caused increased fungal burdens [76]. The immunoprotective role of IL-17 against Candida albicans has also been assessed. Th17 cells have recently been implicated in the defense against the infection, which causes oropharyngeal candidiasis [77]. Based on the model, both Th17 deficient (IL-23p19 −/−) and IL-17R – deficient mice cohorts showed diminished neutrophil recruitment and increased susceptibility to oropharyngeal candidiasis. These data were reinforced in another study examining mice deficient in IL-23 or IL-17A after skin infection with C. albicans compared with wild-type mice or mice deficient in IL-12 or IL-22. This study demonstrated that optimal host defense against cutaneous candidiasis was dependent on IL-23 and IL-17A but not dependent on IL-12 and IL-22 [78].
Insights from IL-17/IL-17R/IL-23 deficiency states in human beings
Linkages between the IL-23/Th17 pathway and susceptibility to infection in human beings have also been reported. In a study of 62 ICU sepsis patients who were assayed for cytokine mRNA levels, a strong relationship was identified between deficient IL-23 expression and the severity of septic infection and mortality [80]. A genetic deficiency of IL-23R is associated with Salmonella infections [81]. The IL-23R pathway is thought to be involved in secondary immunity in these cases because these individuals tend to get recurrent salmonellosis. Several studies have also illustrated the association between IL-12p40 or IL-12Rβ1 deficiency and an increased susceptibility to mycobacteria and Salmonella infections, owing to the inability to mount proper IL-12 and IL-23 cascades [82, 83, 84, 85]. These linkages may be significant in helping to characterize the abnormal IL-23/Th17 pathway seen in psoriasis.
Clinical trials targeting IL-17
Discoveries over the past several decades have shifted our view of psoriasis as primarily a T-cell mediated condition and have paved the way for the development of newer systemic therapies, such as biologics that directly target cytokine signaling or T-cell function. In recent years, IL-12 and IL-23 have become targets for newer biologic therapy agents, such as briakinumab and ustekinumab, which function by targeting the common p40 subunit of IL-12 and IL-23. The extensive information on the effectiveness of anti-p40 antibodies in psoriasis trials [86-91] suggests the importance of the IL-17 pathway in the pathogenesis of psoriasis.
A significant volume of both clinical and experimental data elucidates the effects of systemic therapies on IL-17 expression (Table 3). Whereas these newer biologic agents are generally more specific in the targeting of IL-17 than non-specific conventional immune-modulators and UV radiation therapies, several studies demonstrate the efficacy of systemic psoriasis therapies in reducing IL-17 production. UV exposure, either narrowband ultraviolet B (NB-UVB) or psoralen plus ultraviolet A (PUVA), is effective for psoriasis [92, 93, 94], and two recent clinical trials assessed the influence of phototherapy on the IL-23/Th17 pathway [95, 96]. In one cohort (n=14) of NB-UVB-treated patients, lesion resolution correlated with a reduction in the levels of inflammatory myeloid DCs, T cells, IL-22/IL-17 mRNA expression, and inflammatory products, including IFN-γ, IL-17 and IL-22 [95]. In another study comparing NB-UVB and PUVA, both treatments produced a reduction in PASI, IL-23, TNF-α, IL-22, IL-17, VEGF, and IL-8 [96]. The reductions were particularly large in patients treated with PUVA, which may explain its higher remission rate, as compared with NB-UVB [97]. Follow-up studies with larger cohorts of patients treated with phototherapy to assess its effects on the IL-23/Th17 axis could further strengthen these findings.
Follow-up analysis after conventional systemic therapies has also provided insight in the pathogenesis of psoriasis. In one study, non-lesional and lesional biopsies of patients treated with cyclosporine over 8 weeks were collected and analyzed at weeks 2, 6, and 8 [7]. Fluorescent cell sorting analysis (FACS) revealed significantly more Th17 cells in the psoriatic dermis than in normal skin (P=0.029). Additionally, in these patients, who all achieved PASI 65 or greater, IL-17 and IL-22 mRNA levels were increased in psoriatic skin but normalized with cyclosporine therapy[7].
Biologic treatments offer perhaps the most practical approach for directly targeting the IL17 pathway. In one trial, infliximab therapy decreased the high levels of both Th17 and Th1 cells [35]. Etanercept also reduces IL-17 and IL-22 levels [98, 99, 100, 101]. Two etanercept prospective trials assessed the role of myeloid dendritic cell genes and products and found that IL-17 pathway cytokines were normalized in responders. In a study to determine the extent to which psoriasis genes of responders returned to baseline levels, most cellular markers in the epidermis and dermis did return to non-lesional levels, but IL-17 pathway genes such as IL-17 and IL-22 did not [100]. These results suggest that histological resolution may not be accompanied by full resolution of molecular alterations and residual inflammatory markers may still persist.
One novel agent that directly targets IL-17 is currently undergoing clinical trial. Secukinumab (AIN57), a monoclonal antibody that neutralizes IL-17A, was tested on patients with plaque-type psoriasis (n=36) in a randomized, proof-of-concept clinical trial [102]. Psoriatic lesions improved with rates of adverse reactions, including infection, similar to placebo groups [103]. Dermal IL-17A+ CD3+ T cells and mRNA expression of IL-17 and IL-22 were markedly reduced. These findings suggest that targeted inhibition of IL-17A with secukinumab is a valid therapeutic approach and phase II clinical trials are currently underway. Additional biologic agents, such as LY-24398121, an anti-IL-17 monoclonal antibody, and AMG-827, an anti-IL-17R agent, are also currently being evaluated in phase II clinical trials [104]. Efforts to target a different downstream effector are seen in a phase I clinical trial being completed for an IL-22 antibody, fezakinumab [105]. It is likely that the list of possible indications for altering the IL-23/Th17 pathway may broaden and the number of these agents may increase over time.
Discussion
Considerable progress and new insights into the immunopathogenesis of psoriasis has been made over the past decade. The IL-23/Th17 pathway and its associated proinflammatory molecules, cytokines, and antimicrobial peptides stimulate the amplification of the immune response, leading to the clinical features of psoriasis. IL-23 and IL-17 are two of the key cytokines elevated in psoriatic lesions. This realization has led to the development of strategies to target these specifically as therapeutic options. UVB phototherapy and conventional agents such as cyclosporine also have effects on the IL-17 production. Older biologic therapies that target TNF-α appear to function, at least in part by down-regulating both the Th1 and Th17 pathways. However, in recent years, more specific biological agents that target the Th17 axis, including brakinumab, ustekinumab, and secukinumab have provided powerful new treatment options for psoriasis. Further investigation of the changes in keratinocyte microenvironment, the immune cells of the IL-23/Th17 pathway, including both Th17 and Tc17 cells, and perhaps the relative role of regulatory T cells and possible mechanisms by which they are dysfunctional, will continue to contribute to our understanding of this chronic disease.
References
1. Nair RP, Duffin KC, Helms C, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. Feb 2009;41(2):199-204. [PubMed]2. Blauvelt A. T-helper 17 cells in psoriatic plaques and additional genetic links between IL-23 and psoriasis. J Invest Dermatol. May 2008;128(5):1064-1067. [PubMed]
3. Nickoloff BJ, Qin JZ, Nestle FO. Immunopathogenesis of psoriasis. Clin Rev Allergy Immunol. Oct 2007;33(1-2):45-56. [PubMed]
4. Torti DC, Feldman SR. Interleukin-12, interleukin-23, and psoriasis: current prospects. J Am Acad Dermatol. Dec 2007;57(6):1059-1068. [PubMed]
5. Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. Jun 2009;129(6):1339-1350. [PubMed]
6. Chamian F, Lowes MA, Lin SL, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A. Feb 8 2005;102(6):2075-2080. [PubMed]
7. Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. May 2008;128(5):1207-1211. [PubMed]
8. Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. Dec 2008;226:132-146. [PubMed]
9. Asarch A, Barak O, Loo DS, Gottlieb AB. Th17 cells: a new therapeutic target in inflammatory dermatoses. J Dermatolog Treat. 2008;19(6):318-326. [PubMed]
10. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. Jan 17 2003;278(3):1910-1914. [PubMed]
11. Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. May 2006;116(5):1310-1316. [PubMed]
12. Parham C, Chirica M, Timans J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. Jun 1 2002;168(11):5699-5708. [PubMed]
13. Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. May 2008;8(5):337-348. [PubMed]
14. Wolk K, Witte E, Wallace E, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. May 2006;36(5):1309-1323. [PubMed]
15. Nestle FO, Kaplan DH, Barker J. Psoriasis. New England Journal of Medicine. Jul 30 2009;361(5):496-509. [PubMed]
16. Ma HL, Napierata L, Stedman N, et al. Tumor necrosis factor alpha blockade exacerbates murine psoriasis-like disease by enhancing Th17 function and decreasing expansion of Treg cells. Arthritis Rheum. Feb 2010;62(2):430-440. [PubMed]
17. Watanabe H, Kawaguchi M, Fujishima S, et al. Functional characterization of IL-17F as a selective neutrophil attractant in psoriasis. J Invest Dermatol. Mar 2009;129(3):650-656. [PubMed]
18. Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. Feb 8 2007;445(7128):648-651. [PubMed]
19. Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A. IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J Immunol. Feb 1 2011;186(3):1495-1502. [PubMed]
20. Hedrick MN, Lonsdorf AS, Shirakawa AK, et al. CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. J Clin Invest. Aug 2009;119(8):2317-2329. [PubMed]
21. Kopp T, Lenz P, Bello-Fernandez C, Kastelein RA, Kupper TS, Stingl G. IL-23 production by cosecretion of endogenous p19 and transgenic p40 in keratin 14/p40 transgenic mice: evidence for enhanced cutaneous immunity. J Immunol. Jun 1 2003;170(11):5438-5444. [PubMed]
22. Chan JR, Blumenschein W, Murphy E, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. Nov 27 2006;203(12):2577-2587. [PubMed]
23. Nakajima K, Kanda T, Takaishi M, et al. Distinct Roles of IL-23 and IL-17 in the Development of Psoriasis-Like Lesions in a Mouse Model. J Immunol. Feb 23 2011. [PubMed]
24. Hvid H, Teige I, Kvist PH, Svensson L, Kemp K. TPA induction leads to a Th17-like response in transgenic K14/VEGF mice: a novel in vivo screening model of psoriasis. Int Immunol. Aug 2008;20(8):1097-1106. [PubMed]
25. van der Fits L, van der Wel LI, Laman JD, Prens EP, Verschuren MC. In psoriasis lesional skin the type I interferon signaling pathway is activated, whereas interferon-alpha sensitivity is unaltered. J Invest Dermatol. Jan 2004;122(1):51-60. [PubMed]
26. Tonel G, Conrad C, Laggner U, et al. Cutting edge: A critical functional role for IL-23 in psoriasis. J Immunol. Nov 15 2010;185(10):5688-5691. [PubMed]
27. van der Fits L, Mourits S, Voerman JS, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. May 1 2009;182(9):5836-5845. [PubMed]
28. Liang SC, Tan XY, Luxenberg DP, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. Oct 2 2006;203(10):2271-2279. [PubMed]
29. Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol. Nov 2008;128(11):2625-2630. [PubMed]
30. Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. Sep 2007;8(9):950-957. [PubMed]
31. Boehncke WH, Schon MP. Animal models of psoriasis. Clinics in dermatology. Nov-Dec 2007;25(6):596-605. [PubMed]
32. Nestle FO, Nickoloff BJ. From classical mouse models of psoriasis to a spontaneous xenograft model featuring use of AGR mice. Ernst Schering Research Foundation workshop. 2005(50):203-212. [PubMed]
33. Hansel A, Gunther C, Ingwersen J, et al. Human slan (6-sulfo LacNAc) dendritic cells are inflammatory dermal dendritic cells in psoriasis and drive strong T(h)17/T(h)1 T-cell responses. J Allergy Clin Immunol. Mar 2011;127(3):787-794 e789. [PubMed]
34. Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, et al. Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J Invest Dermatol. Jan 2009;129(1):79-88. [PubMed]
35. Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. May 2010;130(5):1373-1383. [PubMed]
36. Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol. Feb 2009;160(2):319-324. [PubMed]
37. Li J, Chen X, Liu Z, Yue Q, Liu H. Expression of Th17 cytokines in skin lesions of patients with psoriasis. J Huazhong Univ Sci Technolog Med Sci. Jun 2007;27(3):330-332. [PubMed]
38. Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, et al. Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J Immunol. Nov 15 2008;181(10):7420-7427. [PubMed]
39. Takahashi H, Tsuji H, Hashimoto Y, Ishida-Yamamoto A, Iizuka H. Serum cytokines and growth factor levels in Japanese patients with psoriasis. Clin Exp Dermatol. Aug 2010;35(6):645-649. [PubMed]
40. Ortega C, Fernandez AS, Carrillo JM, et al. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol. Aug 2009;86(2):435-443. [PubMed]
41. Res PC, Piskin G, de Boer OJ, et al. Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLoS One. 2010;5(11):e14108. [PubMed]
42. Fan X, Yang S, Huang W, et al. Fine mapping of the psoriasis susceptibility locus PSORS1 supports HLA-C as the susceptibility gene in the Han Chinese population. PLoS Genet. Mar 2008;4(3):e1000038. [PubMed]
43. Nair RP, Stuart PE, Nistor I, et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet. May 2006;78(5):827-851. [PubMed]
44. Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol. Mar 2011;131(3):677-687. [PubMed]
45. Fujishima S, Watanabe H, Kawaguchi M, et al. Involvement of IL-17F via the induction of IL-6 in psoriasis. Archives of dermatological research. Sep 2010;302(7):499-505. [PubMed]
46. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. Feb 14 2003;299(5609):1033-1036. [PubMed]
47. Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol. Sep 1 2009;183(5):3170-3176. [PubMed]
48. Le Borgne M, Etchart N, Goubier A, et al. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity. Feb 2006;24(2):191-201. [PubMed]
49. Varona R, Cadenas V, Gomez L, Martinez AC, Marquez G. CCR6 regulates CD4+ T-cell-mediated acute graft-versus-host disease responses. Blood. Jul 1 2005;106(1):18-26. [PubMed]
50. Homey B, Dieu-Nosjean MC, Wiesenborn A, et al. Up-regulation of macrophage inflammatory protein-3 alpha/CCL20 and CC chemokine receptor 6 in psoriasis. J Immunol. Jun 15 2000;164(12):6621-6632. [PubMed]
51. Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. Sep 2009;129(9):2175-2183. [PubMed]
52. Nograles KE, Zaba LC, Guttman-Yassky E, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. Nov 2008;159(5):1092-1102. [PubMed]
53. Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. Feb 2007;80(2):273-290. [PubMed]
54. Tsunemi Y, Saeki H, Nakamura K, et al. Interleukin-12 p40 gene (IL12B) 3'-untranslated region polymorphism is associated with susceptibility to atopic dermatitis and psoriasis vulgaris. J Dermatol Sci. Nov 2002;30(2):161-166. [PubMed]
55. Safrany E, Szell M, Csongei V, et al. Polymorphisms of the IL23R Gene Are Associated with Psoriasis but not with Immunoglobulin A Nephropathy in a Hungarian Population. Inflammation. Oct 27 2010. [PubMed]
56. Wu Y, Lu Z, Chen Y, Xue F, Chen X, Zheng J. Replication of association between interleukin-23 receptor (IL-23R) and its ligand (IL-12B) polymorphisms and psoriasis in the Chinese Han population. Hum Immunol. Dec 2010;71(12):1255-1258. [PubMed]
57. Dubinsky MC, Wang D, Picornell Y, et al. IL-23 receptor (IL-23R) gene protects against pediatric Crohn's disease. Inflamm Bowel Dis. May 2007;13(5):511-515. [PubMed]
58. McGovern D, Powrie F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut. Oct 2007;56(10):1333-1336. [PubMed]
59. Rueda B, Orozco G, Raya E, et al. The IL23R Arg381Gln non-synonymous polymorphism confers susceptibility to ankylosing spondylitis. Ann Rheum Dis. Oct 2008;67(10):1451-1454. [PubMed]
60. Farago B, Magyari L, Safrany E, et al. Functional variants of interleukin-23 receptor gene confer risk for rheumatoid arthritis but not for systemic sclerosis. Ann Rheum Dis. Feb 2008;67(2):248-250. [PubMed]
61. Huber AK, Jacobson EM, Jazdzewski K, Concepcion ES, Tomer Y. Interleukin (IL)-23 receptor is a major susceptibility gene for Graves' ophthalmopathy: the IL-23/T-helper 17 axis extends to thyroid autoimmunity. J Clin Endocrinol Metab. Mar 2008;93(3):1077-1081. [PubMed]
62. Shibata S, Saeki H, Tsunemi Y, et al. IL-17F single nucleotide polymorphism is not associated with psoriasis vulgaris or atopic dermatitis in the Japanese population. J Dermatol Sci. Feb 2009;53(2):163-165. [PubMed]
63. Ellinghaus E, Ellinghaus D, Stuart PE, et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet. Nov 2010;42(11):991-995. [PubMed]
64. Okamoto Yoshida Y, Umemura M, Yahagi A, et al. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J. Immunol. 2010;184:4414. [PubMed]
65. Teixeira-Coelho M, Cruz A, Carmona J, et al. TLR2 deficiency by compromising p19 (IL-23) expression limits Th 17 cell responses to Mycobacterium tuberculosis. Int Immunol. Feb 2011;23(2):89-96. [PubMed]
66. Luzza F, Parrello T, Monteleone G, et al. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. Nov 1 2000;165(9):5332-5337. [PubMed]
67. Sebkova L, Pellicano A, Monteleone G, et al. Extracellular signal-regulated protein kinase mediates interleukin 17 (IL-17)-induced IL-8 secretion in Helicobacter pylori-infected human gastric epithelial cells. Infect Immun. Sep 2004;72(9):5019-5026. [PubMed]
68. Shiomi S, Toriie A, Imamura S, et al. IL-17 is involved in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Helicobacter. Dec 2008;13(6):518-524. PubMed]
69. Kudva A, Scheller EV, Robinson KM, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. Feb 1 2011;186(3):1666-1674. [PubMed]
70. Raffatellu M, Santos RL, Verhoeven DE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. Apr 2008;14(4):421-428. [PubMed]
71. Kelly MN, Kolls JK, Happel K, et al. Interleukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against Toxoplasma gondii infection. Infect Immun. Jan 2005;73(1):617-621. [PubMed]
72. Guiton R, Vasseur V, Charron S, et al. Interleukin 17 receptor signaling is deleterious during Toxoplasma gondii infection in susceptible BL6 mice. J Infect Dis. Aug 15 2010;202(3):427-435. [PubMed]
73. Kostka SL, Knop J, Konur A, Udey MC, von Stebut E. Distinct roles for IL-1 receptor type I signaling in early versus established Leishmania major infections. J Invest Dermatol. Jul 2006;126(7):1582-1589. [PubMed]
74. Miyazaki Y, Hamano S, Wang S, Shimanoe Y, Iwakura Y, Yoshida H. IL-17 is necessary for host protection against acute-phase Trypanosoma cruzi infection. J Immunol. Jul 15 2010;185(2):1150-1157. [PubMed]
75. Molesworth-Kenyon SJ, Yin R, Oakes JE, Lausch RN. IL-17 receptor signaling influences virus-induced corneal inflammation. J Leukoc Biol. Feb 2008;83(2):401-408. [PubMed]
76. Nigam P, Kwa S, Velu V, Amara RR. Loss of IL-17-producing CD8 T cells during late chronic stage of pathogenic simian immunodeficiency virus infection. J Immunol. Jan 15 2011;186(2):745-753. [PubMed]
77. Rudner XL, Happel KI, Young EA, Shellito JE. Interleukin-23 (IL-23)-IL-17 cytokine axis in murine Pneumocystis carinii infection. Infect Immun. Jun 2007;75(6):3055-3061 [PubMed]
78. Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. Feb 16 2009;206(2):299-311. [PubMed]
79. Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol. Nov 1 2010;185(9):5453-5462. [PubMed]
80. O'Dwyer MJ, Mankan AK, White M, et al. The human response to infection is associated with distinct patterns of interleukin 23 and interleukin 27 expression. Intensive Care Med. Apr 2008;34(4):683-691. [PubMed]
81. Sanal O, Turul T, De Boer T, et al. Presentation of interleukin-12/-23 receptor beta1 deficiency with various clinical symptoms of Salmonella infections. J Clin Immunol. Jan 2006;26(1):1-6. [PubMed]
82. Picard C, Fieschi C, Altare F, et al. Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am J Hum Genet. Feb 2002;70(2):336-348. [PubMed]
83. Fieschi C, Dupuis S, Catherinot E, et al. Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor beta1 deficiency: medical and immunological implications. J Exp Med. Feb 17 2003;197(4):527-535. [PubMed]
84. Pedraza S, Lezana JL, Samarina A, et al. Clinical disease caused by Klebsiella in 2 unrelated patients with interleukin 12 receptor beta1 deficiency. Pediatrics. Oct 2010;126(4):e971-976. [PubMed]
85. de Beaucoudrey L, Samarina A, Bustamante J, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine. Nov 2010;89(6):381-402. [PubMed]
86. Kimball AB, Gordon KB, Langley RG, Menter A, Perdok RJ, Valdes J. Efficacy and safety of ABT-874, a monoclonal anti-interleukin 12/23 antibody, for the treatment of chronic plaque psoriasis: 36-week observation/retreatment and 60-week open-label extension phases of a randomized phase II trial. J Am Acad Dermatol. Feb 2011;64(2):263-274. [PubMed]
87. Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet. May 17 2008;371(9625):1665-1674. [PubMed]
88. Brimhall AK, King LN, Licciardone JC, Jacobe H, Menter A. Safety and efficacy of alefacept, efalizumab, etanercept and infliximab in treating moderate to severe plaque psoriasis: a meta-analysis of randomized controlled trials. Br J Dermatol. Aug 2008;159(2):274-285. [PubMed]
89. Strober BE, Clarke S. Etanercept for the treatment of psoriasis: combination therapy with other modalities. J Drugs Dermatol. May-Jun 2004;3(3):270-272. [PubMed]
90. Reich K, Thaci D, Mrowietz U, Kamps A, Neureither M, Luger T. Efficacy and safety of fumaric acid esters in the long-term treatment of psoriasis--a retrospective study (FUTURE). J Dtsch Dermatol Ges. Jul 2009;7(7):603-611. [PubMed]
91. Kimball AB, Gordon KB, Langley RG, Menter A, Chartash EK, Valdes J. Safety and efficacy of ABT-874, a fully human interleukin 12/23 monoclonal antibody, in the treatment of moderate to severe chronic plaque psoriasis: results of a randomized, placebo-controlled, phase 2 trial. Arch Dermatol. Feb 2008;144(2):200-207.[PubMed]
92. Piskin G, Koomen CW, Picavet D, Bos JD, Teunissen MB. Ultraviolet-B irradiation decreases IFN-gamma and increases IL-4 expression in psoriatic lesional skin in situ and in cultured dermal T cells derived from these lesions. Exp Dermatol. Apr 2003;12(2):172-180. [PubMed]
93. Piskin G, Tursen U, Sylva-Steenland RM, Bos JD, Teunissen MB. Clinical improvement in chronic plaque-type psoriasis lesions after narrow-band UVB therapy is accompanied by a decrease in the expression of IFN-gamma inducers -- IL-12, IL-18 and IL-23. Exp Dermatol. Dec 2004;13(12):764-772. [PubMed]
94. Walters IB, Ozawa M, Cardinale I, et al. Narrowband (312-nm) UV-B suppresses interferon gamma and interleukin (IL) 12 and increases IL-4 transcripts: differential regulation of cytokines at the single-cell level. Arch Dermatol. Feb 2003;139(2):155-161. [PubMed]
95. Johnson-Huang LM, Suarez-Farinas M, Sullivan-Whalen M, Gilleaudeau P, Krueger JG, Lowes MA. Effective narrow-band UVB radiation therapy suppresses the IL-23/IL-17 axis in normalized psoriasis plaques. J Invest Dermatol. Nov 2010;130(11):2654-2663. [PubMed]
96. Coimbra S, Oliveira H, Reis F, et al. Interleukin (IL)-22, IL-17, IL-23, IL-8, vascular endothelial growth factor and tumour necrosis factor-alpha levels in patients with psoriasis before, during and after psoralen-ultraviolet A and narrowband ultraviolet B therapy. Br J Dermatol. Dec 2010;163(6):1282-1290. [PubMed]
97. Karrer S, Eholzer C, Ackermann G, Landthaler M, Szeimies RM. Phototherapy of psoriasis: comparative experience of different phototherapeutic approaches. Dermatology. 2001;202(2):108-115. [PubMed]
98. Caproni M, Antiga E, Melani L, Volpi W, Del Bianco E, Fabbri P. Serum levels of IL-17 and IL-22 are reduced by etanercept, but not by acitretin, in patients with psoriasis: a randomized-controlled trial. J Clin Immunol. Mar 2009;29(2):210-214. [PubMed]
99. Zaba LC, Suarez-Farinas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. Nov 2009;124(5):1022-1010 e1021-1395. [PubMed]
100. Suarez-Farinas M, Fuentes-Duculan J, Lowes MA, Krueger JG. Resolved psoriasis lesions retain expression of a subset of disease-related genes. J Invest Dermatol. Feb 2011;131(2):391-400. [PubMed]
101. Zaba LC, Cardinale I, Gilleaudeau P, et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. Dec 24 2007;204(13):3183-3194. [PubMed]
102. Kurzeja M, Rudnicka L, Olszewska M. New interleukin-23 pathway inhibitors in dermatology: ustekinumab, briakinumab, and secukinumab. Am J Clin Dermatol. Apr 1 2011;12(2):113-125. [PubMed]
103. Hueber W, Patel DD, Dryja T, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. Oct 6 2010;2(52):52ra72. [PubMed]
104. Garber K. Immunology's quiet upheaval. Nat Biotechnol. Aug 2009;27(8):687-689. [PubMed]
105. National Psoriasis Foundation. Drugs in Development http://www.psoriasis.org/page.aspx?pid=664. Accessed October, 2011.
106. Lillis JV, Guo CS, Lee JJ, Blauvelt A. Increased IL-23 expression in palmoplantar psoriasis and hyperkeratotic hand dermatitis. Arch Dermatol. Aug 2010;146(8):918-919. [PubMed]
107. Lee E, Trepicchio WL, Oestreicher JL, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. Jan 5 2004;199(1):125-130. [PubMed]
108. Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. Oct 1998;111(4):645-649. [PubMed]
109. Grossman RM, Krueger J, Yourish D, et al. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc Natl Acad Sci U S A. Aug 1989;86(16):6367-6371. [PubMed]
110. Neuner P, Urbanski A, Trautinger F, et al. Increased IL-6 production by monocytes and keratinocytes in patients with psoriasis. J Invest Dermatol. Jul 1991;97(1):27-33. [PubMed]
111. Umemura M, Yahagi A, Hamada S, et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J Immunol. Mar 15 2007;178(6):3786-3796. [PubMed]
© 2012 Dermatology Online Journal