Identification of a new mutation in the gene coding for hairless protein responsible for alopecia universalis: The importance of direct gene sequencing
Published Web Location
https://doi.org/10.5070/D32t39d9d8Main Content
Identification of a new mutation in the gene coding for hairless protein responsible for alopecia universalis: The importance
of direct gene sequencing
Stefania Nucara MA, Emma Colao MD, Graziella Mangone MD, Francesco Baudi MD, Fernanda Fabiani MA, Donatella Nocera MA, Giuseppe
Passafaro MA, Teresa Longo MA, Anna Elisa Laria MA, Paola Malatesta MA, Rosario Amato MD PhD, Francesco Trapasso MD PhD, Nicola
Perrotti MD
Dermatology Online Journal 17 (1): 3
Unit of Genetic PathologyUniversity Hospital Policlinico Mater Domini
Department of Experimental and Clinical Medicine, University of Catanzaro
Viale Salvatore Venuta, Catanzaro, Italy.
Abstract
Mutations in the gene HR coding for the hairless protein are associated with atrichia with papular lesions (APL), an autosomal recessive form of alopecia universalis that is characterized by generalized scalp and body atrichia with papular lesions. We here describe a South Italian family of ancient Albanian heritage. The full phenotype with complete atrichia was expressed in 2 siblings, whereas the parents and one sister were unaffected. Direct sequencing of the gene coding for the hairless protein allowed the identification of a new mutation in exon 17. Consistent with the recessive inheritance of the disease, both the siblings were homozygous for the mutation, whereas the parents and the unaffected sister where heterozygous. A relevant discrepancy with a haplotype linkage study is reported, stressing the importance of gene sequencing in genetic diagnosis and counseling because linkage studies can be biased by recombination events.
Introduction
Mutations in the gene Hr coding for the Hairless protein are associated with atrichia with papular lesions (APL), an autosomal recessive form of alopecia universalis that is characterized by generalized scalp and body atrichia with papular lesions [1].
The HR protein belongs to the family of nuclear repressor co-repressors. Most of the nuclear receptors act as repressors of transcription in the absence of the specific ligand. It is now known that alterations in the transcriptional repression activity of nuclear receptors are important in the development of human disease. Repression by nuclear receptors occurs through their association with specific proteins that include co-repressors that are part of a large multi-protein complex that functions via associated enzymatic activities that modify chromatin structure, such as histone deacetylation [2].
The Hr gene is regulated at the transcriptional level by the thyroid hormone receptor (TR) [3]. The gene product HR is located in the nucleus and has been shown to interact with the thyroid hormone receptor [3, 4], histone deacetylase (HDAC) [5, 6] and the vitamin D receptor VDR [7].
There is evidence suggesting that HR protein acts as a co-repressor for thyroid hormone and vitamin D receptors [4, 5, 7]. In particular it is believed that HR functions as a co-repressor of VDR to block 1,25(OH)2D3 action on keratinocytes [8]. It has been hypothesized that HR, VDR, and TR converge to control a common pathway in hair cycling (Figure 1A), probably by assembling a transcriptional complex that, recruiting HDAC, represses the expression of Wise, an inhibitor of Wnt signaling. According to this model, HR controls the timing of Wnt signaling required for hair cycling and stem cell differentiation [9]. A proper interaction with VDR seems to be essential for normal hair growth, in fact mutations in either the hairless gene or the vitamin D receptor gene result in similar phenotypes [10]. On the other hand the role of the gene coding for the thyroid hormone receptor needs to be better defined because mutations in this gene that disrupt the interaction between HR protein and TR, are not responsible for atrichia with papular lesions [11].
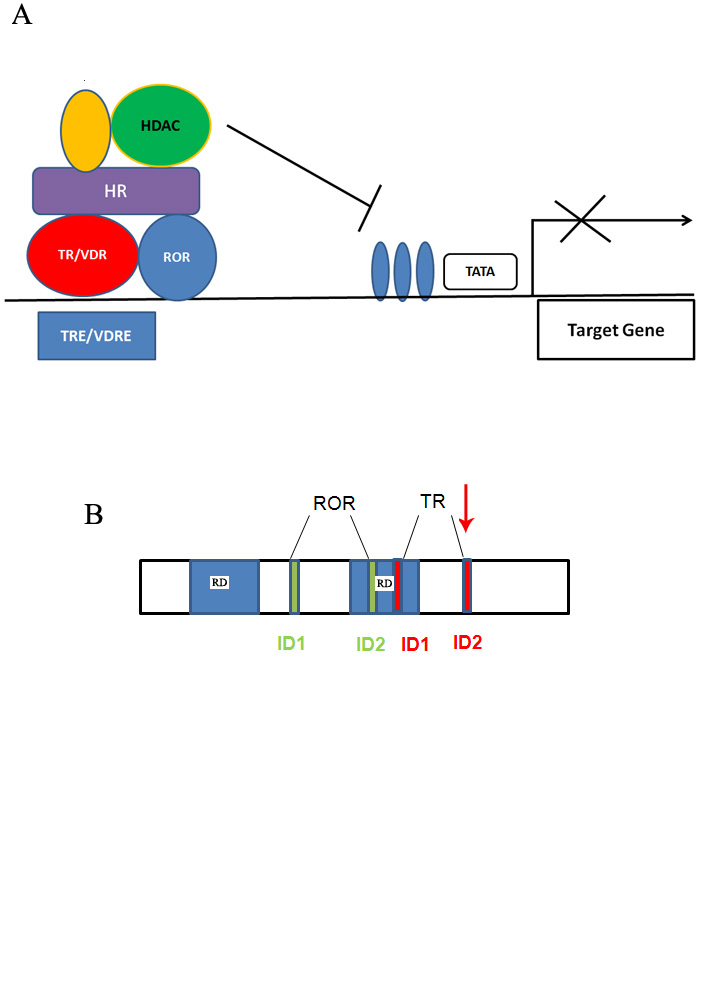 |
| Figure 1 |
|---|
| Figure 1A. Schematic of the interaction of the HR protein with transcriptional regulators: Thyroid hormone receptor (TR),
Retinoic acid related orphan receptor (ROR) and Histone deacetylase (HDAC). Figure 1B. Schematic of the domains in HR protein: repression domains (RD), interaction domains 1 and 2 (ID 1 and ID 2) for retinoic acid related orphan receptor (ROR, green) and thyroid hormone receptor (TR, red). The red arrows shows the site of the mutation in the patient described here. |
In humans, two major HR isoforms are expressed because of differential splicing of exon 17 [12, 13]. HR isoform-a encodes the full-length HR, and isoform-b encodes an HR protein with a 55-amino acid deletion, HR_1072-1126 [14]. Keratinocytes are thought to be the major site for HR-VDR interactions in the regulation of the hair cycle and were shown to express the full-length HR [8].
Both the full-length HR isoform-a and HR_1072-1126 isoform b interact with the VDR, but only the full-length HR is able to repress VDR dependent transactivation [14]. The role of the c-terminal tail of the protein needs to be better defined because the amino acids 980-1084 have been demonstrated to be unable to bind VDR and to repress VDR dependent transactivation [7]. Figure 1B depicts the regions of interaction between the hairless protein and the receptors.
Several types of disease-causing Hr mutations have been identified in APL families, including nonsense, missense, insertion, and deletion mutations. Most mutations have been noted in the homozygous state in consanguineous families [15].
Case report
We here present the case of a family with alopecia universalis.
Genetic counseling for rare disorders is sometimes integrated by linkage analysis that can provide information about the mechanisms of heredity as well as about the chromosomal localization of the affected gene. Unfortunately, linkage studies can be biased by recombination events that can cause loss of association.
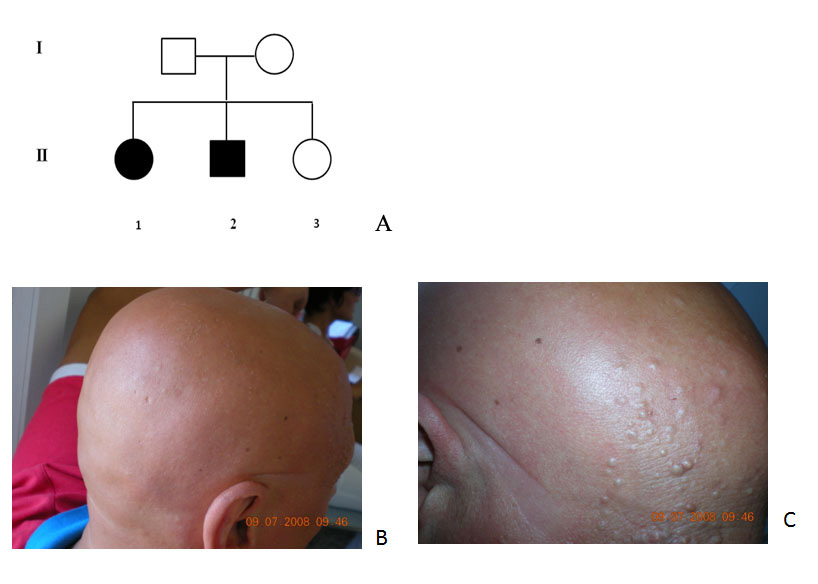 |
| Figure 2 |
|---|
| Figure 2A. Two generations pedigree with the proband (II-1), the affected sibiling (II-2), and the unaffected sibling (II-3). Figure 2B. Clinical presentation of the proband with APL with complete alopecia of the scalp. Figure 2C. Note the papules at a closer view. |
The proband, a 35-year-old Italian female from ancient Albanian descent, born from non-consanguineous parents (Figure 2A) shows the complete phenotype associated with atrichia with papular lesions (Figure 2B) (APL; OMIM #209500). A closer view of the skin in the proband discloses the typical papular lesions (Figure 2C). A male sibling shows an identical condition, whereas the parents and one female sibling are unaffected (Figure 2A). Except for the affected sibling there is no family history of APL. At birth, the patient and the affected sibling presented with patches of hair that shed and never re-grew. They currently have generalized scalp and body alopecia. A papular eruption is present in different areas of the skin both in the proband and the affected sibling. A scalp biopsy was proposed but was firmly denied because it was considered to be a traumatic procedure. On the other hand, although histological assessment forms part of the diagnostic criteria, a scalp biopsy is not always feasible. Based on the current literature, the papules are made up of follicular cysts filled with cornified material [16].
The family had previously sought the advice of a genetic consultant who, based on linkage analysis of polymorphic markers on the short arm of chromosome 8, had established a linkage with the region of chromosome 8 that includes the hairless gene.
The inheritance was consistent with a recessive model. The proband and the affected brother were found to be homozygous for the haplotype linked with the hairless gene. Both the parents were found to be heterozygous, whereas the healthy sister was found to be homozygous for the haplotype not linked with the gene. Based on these results the healthy sister was completely reassured that she did not carry the risk genotype.
To identify the underlying mutation in the HR gene responsible for the APL phenotype in the patient, all exons and splice junctions were PCR amplified from genomic DNA and sequenced directly in an ABI Prism 310 Automated Sequencer, using the ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems), following purification with Centriflex Gel Filtration Cartridges (Edge Biosystems, Gaithersburg, MD, USA). We first PCR amplified and directly sequenced exon 9 of Hr in order to screen for the most common 2147delC mutation. Because our proband did not carry the common mutation 2147delC, we subsequently amplified and sequenced the remaining exons of Hr. Sequencing of the PCR product corresponding to exon 17 revealed a homozygous frameshift mutation due to the insertion of four bases (GGCC) at the 5' end of exon 17, codon 1075.
Interestingly the affected brother was found to be homozygous for the mutation, whereas the parents and the healthy sister were all heterozygous.
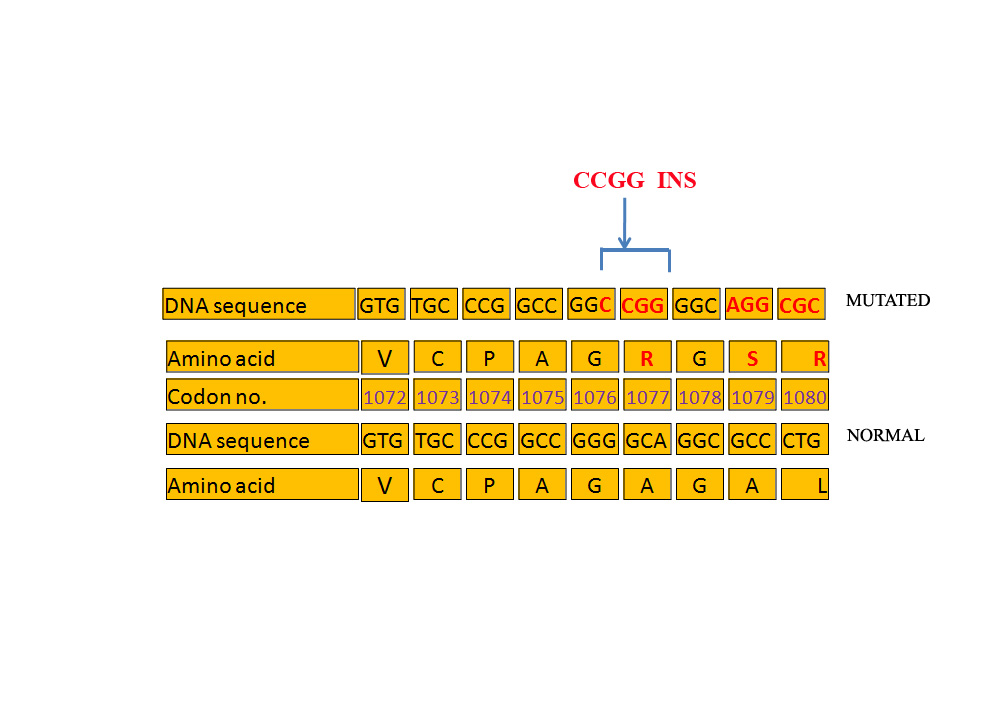 |
| Figure 3 |
|---|
| Figure 3. Schematic of the mutation described in the DNA sequence |
The frameshift mutation disrupts the coding sequence of exon 17 and destroys the second interaction domain for TR (TR-ID2; 980-1084), 37 bases downstream to the C terminal essential residues I/L-I-X-X-L/V-V (1026-1038) similarly to those identified for N-CoR and SMRT binding to TR and RAR [2] (Figure 1B and Figure 3). The mutation, besides disrupting the normal sequence, also introduces a stop at codon 1145, 31 bases upstream from the missense mutation Gln1176Term [17] that interrupts the coding sequence at codon 1176 and is also associated with atrichia with papular lesions.
In terms of counseling, full sequencing of the gene helped to define correctly the genotype of the unaffected sister, a result that can be of significant value in genetic counseling, especially in small communities characterized by a significant inbreeding.
References
1. Ahmad, W.; Irvine, A. D.; Lam, H.; Buckley, C.; Bingham, E. A.; Panteleyev, A. A.; Ahmad, M.; McGrath, J. A.; Christiano, A. M. (1998). A missense mutation in the zinc-finger domain of the human hairless gene underlies congenital atrichia in a family of Irish travellers. Am. J. Hum. Genet. 63: 984-991. [PubMed]2. Thompson C. C. (2009) Hairless is a nuclear receptor corepressor essential for skin function. Nuclear Receptor Signaling ( NRS) ( 7) 1 – 11. [PubMed]
3. Thompson C. Bottcher M.C. (1997) The product of a thyroid hormone-responsive gene interacts with thyroid hormone receptors Proc. Natl. Acad. Sci. USA (94) 8527–8532. [PubMed]
4. Potter GB, Beaudoin GM 3rd, DeRenzo CL, Zarach JM, Chen SH, Thompson CC. (2001) The hairless gene mutated in congenital hair loss disorders encodes a novel nuclear receptor corepressor. Genes Dev. (20) 2687-701. [PubMed]
5. Potter GB, Zarach JM, Sisk JM, Thompson CC. (2002) The thyroid hormone-regulated corepressor hairless associates with histone deacetylases in neonatal rat brain. Mol Endocrinol. (11):2547-60. [PubMed]
6. Djabali K, Christiano AM. (2004) Hairless contains a novel nuclear matrix targeting signal and associates with histone deacetylase 3 in nuclear speckles. Differentiation. (8):410-8. [PubMed]
7. Hsieh JC, Sisk JM, Jurutka PW, Haussler CA, Slater SA, Haussler MR, Thompson CC (2003). Physical and Functional Interaction between the Vitamin D Receptor and Hairless Corepressor, Two Proteins Required for Hair Cycling. J.Biol.Chem. (278):. 38665–38674. [PubMed]
8. Xie Z., Chang S., Oda Y., Bikle D.D. (2006) Hairless Suppresses Vitamin D Receptor Transactivation in Human Keratinocytes. Endocrinology (147):314–323. [PubMed]
9. Thompson CC, Sisk JM, Beaudoin GM 3rd (2006) Hairless and Wnt signaling: allies in epithelial stem cell differentiation. Cell Cycle. (17):1913-7. [PubMed]
10. Miller J, Djabali K, Chen T, Liu Y, Ioffreda M, Lyle S, Christiano AM, Holick M, Cotsarelis G. (2001) Atrichia caused by mutations in the vitamin D receptor gene is a phenocopy of generalized atrichia caused by mutations in the hairless gene.J. Invest Dermatol. (117):612-7. [PubMed]
11. Djabali K, Zlotogorski A, Metzker A, Ben-Amitai D, Christiano AM. (2004). Interaction of hairless and thyroid hormone receptor is not involved in the pathogenesis of atrichia with papular lesion. Exp Dermatol. (13):251-6. [PubMed]
12. Cichon S, Anker M, Vogt IR, Rohleder H, Putzstuck M, Hillmer A,Farooq SA, Al-Dhafri KS, Ahmad M, Haque S, Rietschel M, Propping P, Kruse R, NothenMM (1998) Cloning, genomic organization, alternative transcripts and mutational analysis of the gene responsible for autosomal recessive universal congenital alopecia. Hum Mol Genet (7):1671–1679. [PubMed]
13. Ahmad W, Zlotogorski A, Panteleyev AA, Lam H, Ahmad M, Haque MF, Abdallah HM, Dragan L, Christiano AM (1999). Genomic organization of the human hairless gene (HR) and identification of a mutation underlying congenital atrichia in an Arab Palestinian family. Genomics (56):141–148. [PubMed]
14. Malloy P. J., Wang J., Jensen K., Feldman D. (2009).Modulation of Vitamin D Receptor Activity by the Corepressor Hairless: Differential Effects of Hairless Isoforms. Endocrinology. (150) :4950–4957. [PubMed]
15. Paradisi M, Massé M, Martinez-Mir A, Lam H, Pedicelli C, Christiano AM (2005) Identification of a novel splice site mutation in the human hairless gene underlying atrichiawith papular lesions. Eur J Dermatol. (15): 332-8. [PubMed]
16. Yip L, Horev L, Sinclair R, Zlotogorski A (2008). Atrichia with Papular Lesions: A Report of Three Novel Human Hairless Gene Mutations and a Revision of Diagnostic Criteria. Acta Derm Venereol (88): 346–349
17. Henn W, Zlotogorski A, Lam H, Martinez-Mir A, Zaun H, Christiano AM.( 2002) Atrichia with papular lesions resulting from compound heterozygous mutations in the hairless gene: A lesson for differential diagnosis of alopecia universalis. J Am Acad Dermatol. (47):519-23. [PubMed]
© 2011 Dermatology Online Journal