Vogt-Koyanagi-Harada syndrome: Association with alopecia areata
Published Web Location
https://doi.org/10.5070/D32qz018fcMain Content
Vogt-Koyanagi-Harada syndrome: Association with alopecia areata
Waqar M Haque MD, Mohsin R Mir MD, Sylvia Hsu MD
Dermatology Online Journal 15 (12): 10
Department of Dermatology, Baylor College of Medicine, Houston, Texas. shsu@bcm.eduAbstract
Vogt-Koyanagi-Harada (VKH) syndrome is an inflammatory condition characterized by bilateral uveitis, vitiligo, poliosis, alopecia, and dysacousia. The syndrome results from a T cell mediated autoimmune attack on melanocytes in genetically susceptible individuals. We present a case of VKH syndrome and propose that the alopecia and poliosis described in the original reports by ophthalmologists could instead be alopecia areata with re-growth of white hair.
Case report
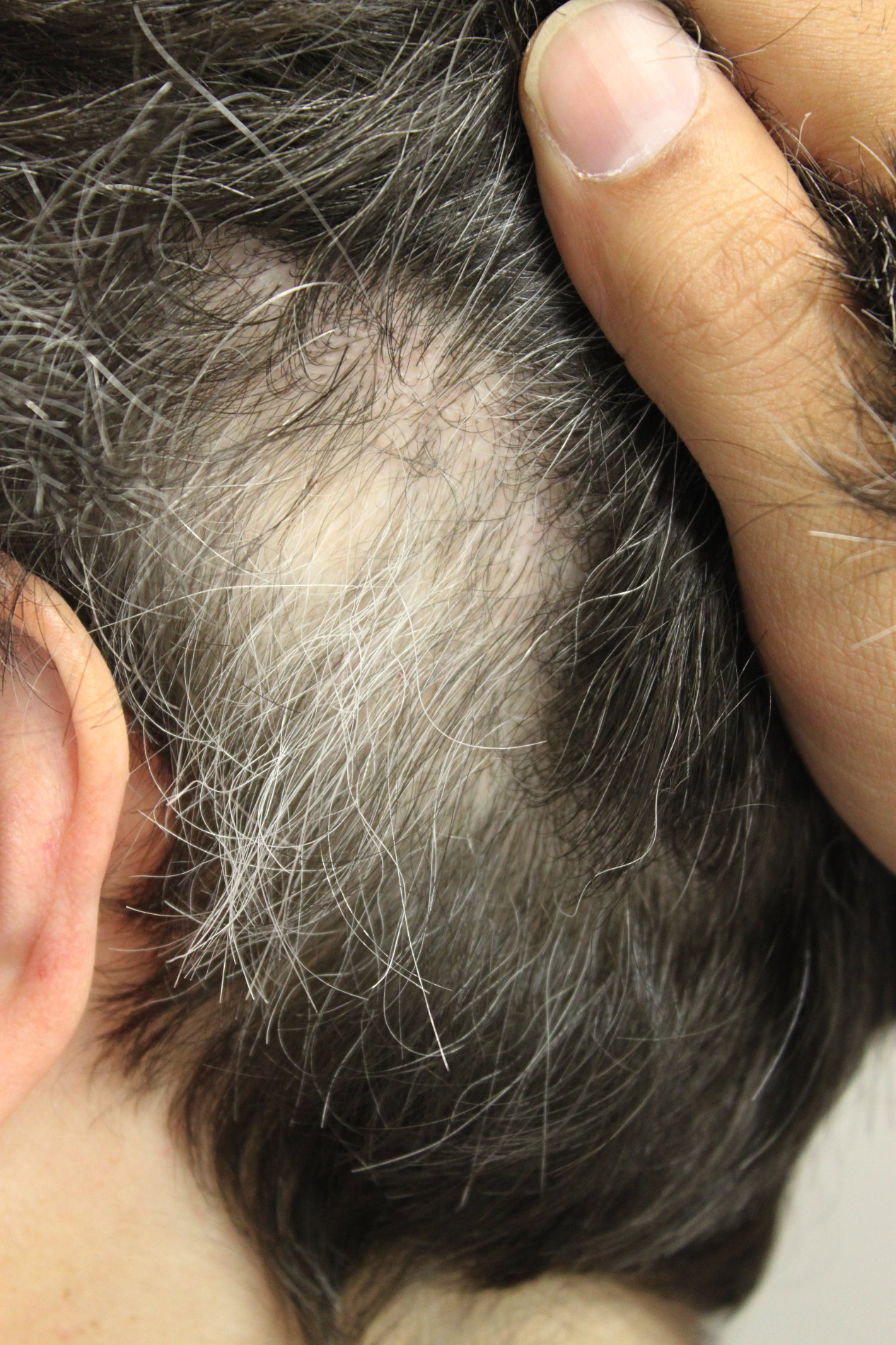 |
| Figure 1 |
|---|
| Figure 1. Well-circumscribed patch of alopecia with re-growth of white hair |
A 42-year-old Caucasian woman was referred by her ophthalmologist for evaluation of alopecia. She reported a 4-year history of alopecia characterized by well-circumscribed patches of hair loss. The hair loss was rapid and asymptomatic, and re-growth of hair was spontaneous. She noted that the hairs that grew back were completely white.
Physical exam revealed well-circumscribed patches of alopecia with varying degrees of re-growth. Although she had some graying of her normal hair, the hairs within the patches of alopecia were strikingly non-pigmented (Fig. 1). Subtle, depigmented patches of skin were observed bilaterally around the eyebrows and upper eyelids.
She reported a history of idiopathic, bilateral uveitis for which she had closely been followed by her ophthalmologist since 2002. Her disease required long-term systemic immunosuppressive medications and eventually vitreous surgery for her extensive macular damage. Upon further questioning, she experienced a febrile illness with meningismus, severe headaches, and dysacousia several weeks prior to the onset of her uveitis in 2002. This constellation of findings is diagnostic for Vogt-Koyanagi-Harada (VKH) syndrome.
Discussion
Vogt-Koyanagi-Harada syndrome is a multisystem autoimmune disorder affecting pigmented cells in the skin, eyes, ears, and central nervous system. The diagnosis is based on a clinical constellation of symptoms involving these organ systems. Vogt, a Swiss ophthalmologist, reported a single case in 1906 [1]. In 1926 Harada, a Japanese ophthalmologist, reported another case, and in 1929, Koyanagi, another Japanese ophthalmologist, reported 6 more cases [2, 3]. In 1932 Babel concluded that all these patients had the same disorder and coined the term Vogt-Koyanagi syndrome, which we now know as Vogt-Koyanagi-Harada syndrome [4]. The disease primarily affects races with darker pigmentation including Asians, Hispanics, and Middle Easterners, but is less common among individuals of African descent [5]. However, in a series of 46 patients, Nussenblatt and Palestine reported that 50 percent of their patients classified themselves as being Caucasian and 35 percent black [6]. Vogt-Koyanagi-Harada syndrome is more common in women and usually occurs during the 3rd or 4th decade of life.
Although the exact etiology of VKH syndrome remains uncertain, numerous studies support a T cell mediated autoimmune response against melanocytes. Human leukocyte antigen (HLA) studies suggest that genetic predisposition plays a role in the pathogenesis of VKH syndrome. Patients expressing the HLA-DRB1*0405 allele are at the highest risk, while HLA-DR4 and HLA-Dw53 are also implicated [7]. Tyrosinase and tyrosinase-related proteins had been implicated as the target antigens on melanocytes; however investigations failed to demonstrate increased risk of disease based on mutations in the tyrosinase gene family [8]. In a recent study, Otani et al. identified a novel auto-antigen (KU-MEL-1) that may be the target in VKH syndrome [9]. The inciting event that triggers an autoimmune response in susceptible individuals has yet to be identified.
The clinical manifestations of VKH syndrome occur in phases. In the initial prodromal or meningoencephalitic phase, patients experience fever, headache, meningismus, vertigo, and dysacusis. Recovery is usually complete. Days to weeks later, the uveitic phase ensues. Patients may have anterior and/or posterior uveitis, which clinically presents with blurred vision, photophobia, and ocular pain. A swollen and hyperemic optic nerve may be seen in 87 percent of patients at this time [10]. Weeks to months later as the uveitis improves, the convalescent phase begins. Integumentary involvement, including vitiligo, alopecia, and poliosis affecting mainly the head and neck, occurs during this period. The chronic phase of VKH syndrome is characterized by recurrence of ocular complications, such as choroidal neovasular membranes, hemorrhage, cataracts, and subretinal fibrosis [4].
Vogt-Koyanagi-Harada syndrome is diagnosed by the clinical presence of uveitis as demonstrated by fluorescein angiography, indocyanine green angiography, or ultrasonography of the eyes [11]. There are 5 criteria that divide VKH syndrome into complete, incomplete, and probable categories on the basis of the clinical characteristics of the patient [12]. The criteria are as follows: 1) absence of penetrating trauma; 2) lack of systemic disease, which could also explain the findings; 3) presence of bilateral ocular pathology; 4) neurologic/auditory findings including tinnitus or meningismus; 5) presence of integumentary findings such as alopecia, poliosis, or vitiligo. Incomplete VKH syndrome is defined as disease that fulfills the first 3 diagnostic criteria as well as either the 4th or 5th criterion. When only the first 3 criteria are met, patients are considered to have probable disease.
The present case describes a patient with well-circumscribed patches of hair loss followed by re-growth of non-pigmented white hair, clinically characteristic for alopecia areata. To our knowledge, only one previous case report has suggested a link between alopecia areata and VKH syndrome, also describing a woman who had alopecia followed by re-growth of non-pigmented hair [13]. The histologic features in that case, including increased ratio of telogen/ catagen:anagen follicles and peribulbar mononuclear infiltrate, were consistent with alopecia areata.
The association of alopecia areata with VKH syndrome would be in accordance with the pathogenesis of the 2 conditions. Alopecia areata is also a T cell mediated autoimmune disorder which targets follicular antigens of possibly keratinoctye or melanocyte origin [14]. HLA-DR4, DR11, and DQ7 are associated with severe cases with early onset. Alopecia areata can be associated with multiple other autoimmune diseases including pernicious anemia, lupus erythematosus, myasthenia gravis, celiac disease, thyroid disease, and vitiligo. We suggest that the alopecia with poliosis in the original ophthalmology literature could instead be alopecia areata with re-growth of white hair. Most published reports of this uncommon disorder are in the ophthalmology literature, so it is possible that the clinical manifestations of alopecia areata were described as alopecia with poliosis. Further investigation is warranted to elucidate the triggering events and detailed pathogenesis of both disorders.
References
1. Vogt A. Frühzeitiges Ergrauen der Zilien und Bemerkungen üben den sogannten plözlichen Eintritt dieser Veränderung. Klin Monatsbl Augenheilkd 1906; 44: 228-242.2. Harada E. Clinical observations of nonsuppurative choroiditis. Acta Soc Ophthalmol Jpn 1926; 30: 356.
3. Koyanagi Y. Dysakusis, alopecia un poliosis bei schwerer uveitis nicht traumatischen ursprungs. Klin Monatsbl Augenheilkd 1929; 82: 194-211.
4. Babel J. Syndrome de Vogt-Koyanagi. Schweiz Med wochenschr 1932; 44: 1136-1140.
5. Andreoli CM, Fostor CS. Vogt-Koyanagi-Harada Disease. Int Ophthal Clinic 2006 Spring; 46 (2): 111-22. [PubMed]
6. Nussenblatt RB, Palestine AG. The Vogt-Koyanagi-Harada Syndrome In: Uveitis Fundamentals and Clinical Practice. Chicago: Year Book Medical Publishers, 1989: 274-290.
7. Shindo Y, Inoko H, Yamamoto T, et al. HLA-DRB1 typing of Vogt-Koyanagi 'Harada's disease by PCR-RFLP and the strong association with DRB1*0405 and DRB1*0410. Br J Opthalmol 1994 Mar; 78 (3): 223-6. [PubMed]
8. Horie Y, Takemoto Y, Miyazaki A, Namba K, Kase S, Yoshida K, Ota M, Hasumi Y, Inoko H, Mizuki N, Ohno S. Tyrosinase gene family and Vogt-Koyanagi-Harada disease in Japanese patients. Mol Vis. 2006 Dec 20;12:1601-5. [PubMed]
9. Otani S, Sakurai T, Yamamoto K, Fujita T, Matsuzaki Y, Goto Y, Ando Y, Suzuki S, Usui M, Takeuchi M, Kawakami Y. Frequent immune response to a melanocyte specific protein KU-MEL-1 in patients with Vogt-Koyanagi-Harada disease. Br J Ophthalmol. 2006 Jun; 90(6):773-7. [PubMed]
10. Ohno S, Minakawa R, Matsuda H. Clinical studies of Vogt-Koyanagi-Harada's disease. Jpn J Ophthalmol 1988; 32 (3): 334-43. [PubMed]
11. Forster DJ, Cano MR, Green RL, et al. Echographic features of the Vogt-Koyanagi-Harada syndrome. Arch Ophthalmol 1990 Oct; 108 (10): 1421-6. [PubMed]
12. Read RW, Holland GN, Rao NA, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001 May; 131 (5): 647-52. [PubMed]
13. DiPreta EA, Smith KJ, Williams J, Skelton AH. Histopathologic findings in the alopecia associated with Vogt-Koyanagi-Harada disease. J Cutan Med Surg 2000 Jul; 4 (3): 156-60. [PubMed]
14. Wasserman D, Guzman-Sanchez DA, Scott K, McMichael A. Alopecia areata. Int J Dermatol 2007 Feb; 46 (2): 121-31. [PubMed]
© 2009 Dermatology Online Journal