Fatal ALK-negative systemic anaplastic large cell lymphoma presenting with disseminated cutaneous dome-shaped papules and nodules
Published Web Location
https://doi.org/10.5070/D32b5993kbMain Content
Fatal ALK-negative systemic anaplastic large cell lymphoma presenting with disseminated cutaneous dome-shaped papules and
nodules
Jayne Eleanor Bird MD, Justin John Leitenberger MD, Alvin Solomon MD, Andrew Blauvelt MD, Sam Hopkins MD
Dermatology Online Journal 18 (5): 5
Oregon Health and Science University, Portland, OregonAbstract
Anaplastic large cell lymphoma (ALCL) is classified as a CD30 positive non-Hodgkin lymphoma. Systemic ALCL (S-ALCL) is further subdivided into two subgroups based on anaplastic lymphoma kinase (ALK) expression. In systemic ALCL, positive ALK expression correlates with a favorable prognosis, whereas negative ALK expression correlates with poorer overall survival. By definition, primary cutaneous ALCL (cut-ALCL) is limited to the skin and is uniformly ALK-negative. Cut-ALCL closely resembles LyP with regards to its benign clinical course and CD30 positivity. We describe a unique case of ALK-negative (ALK-) S-ALCL presenting with cutaneous disseminated dome-shaped papules.
Case synopsis
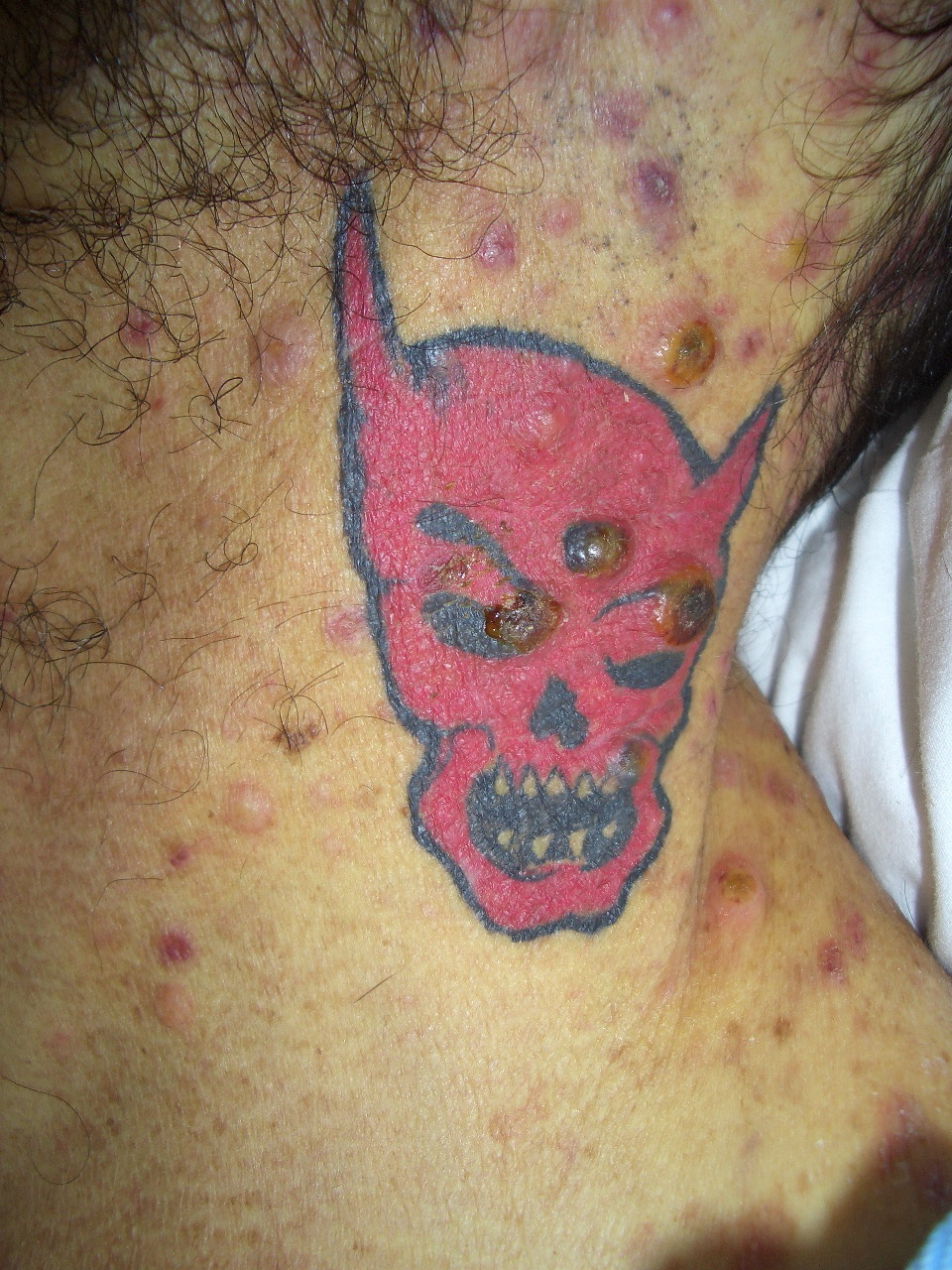 | 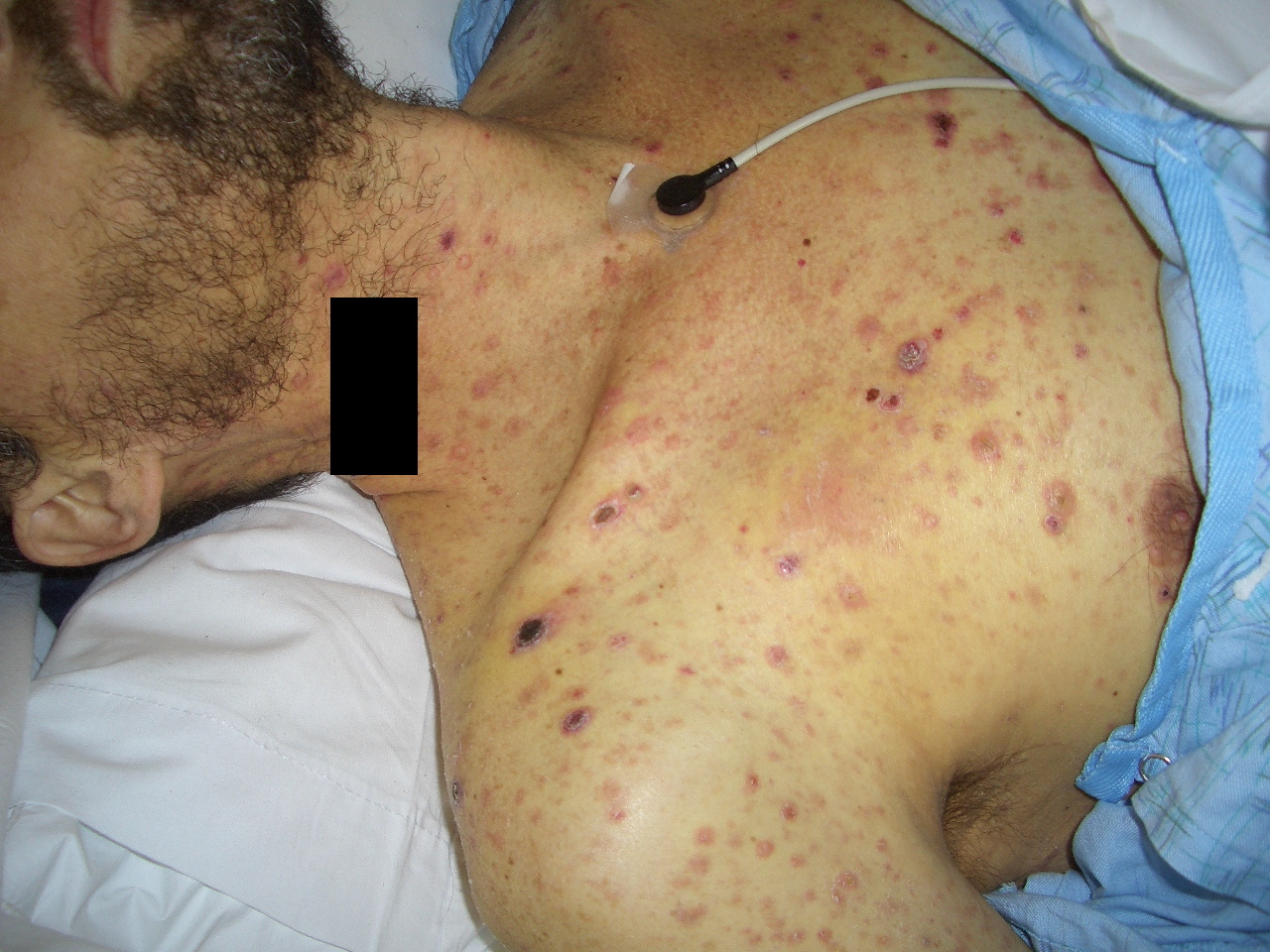 |
| Figure 1A | Figure 1B |
|---|---|
| Figures 1A and 1B. Erythematous, dome-shaped papules with central crust on the neck, torso, and upper extremities. | |
A previously healthy 47-year-old male presented with hepatitis, pancytopenia, altered mental status, and disseminated dome-shaped violaceous and brown papules and nodules, many with central umbilication and crust. The papules and nodules were in different stages of evolution (Figures 1A and 1B) and had appeared approximately two months after a previous hospital admission for altered mental status and un-explained hepatitis. Clinically palpable lymphadenopathy was absent. A skin biopsy revealed sheets of large pleomorphic cells infiltrating the dermis with necrosis of the epidermis (Figure 2A).
 | 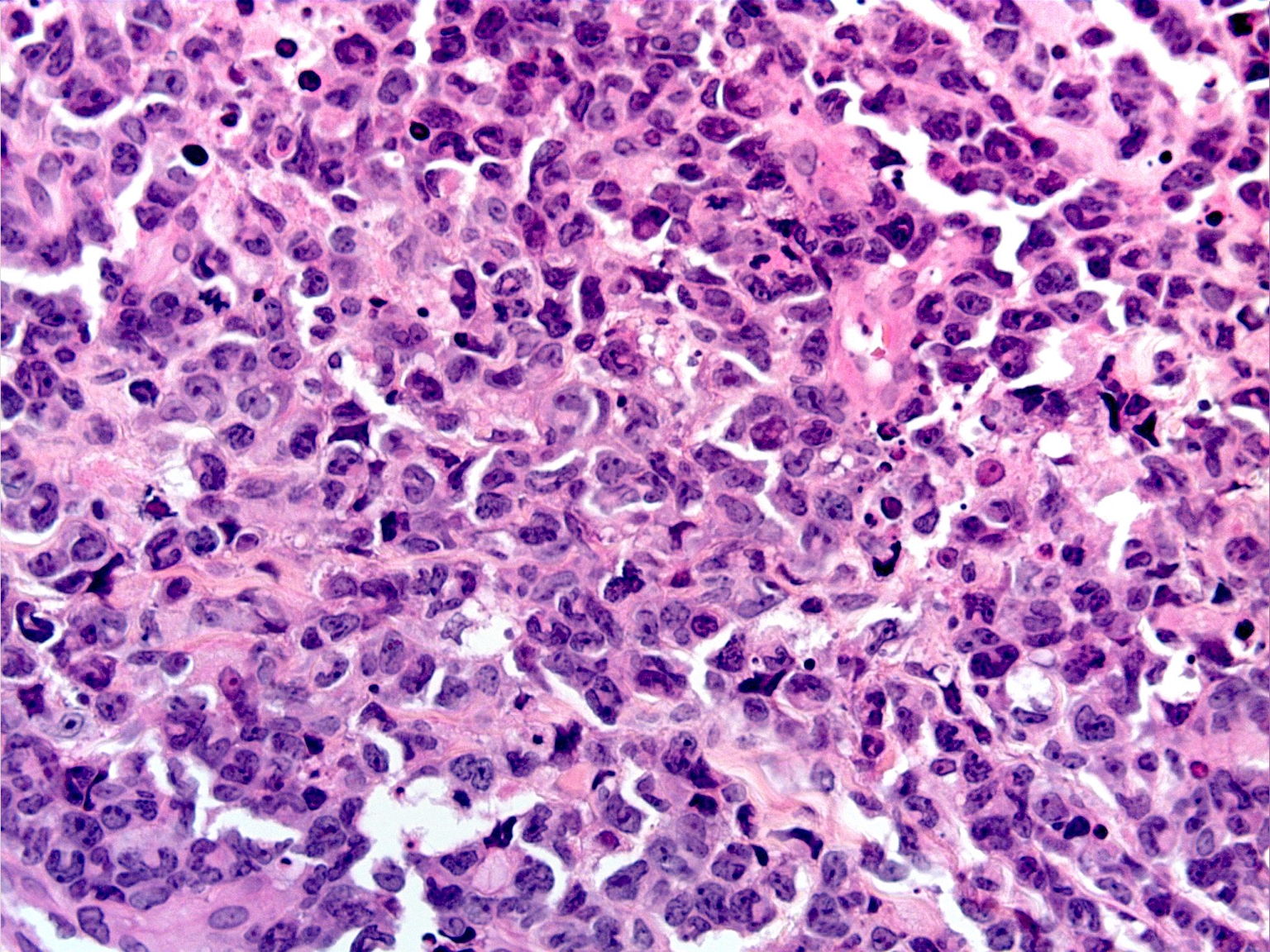 |
| Figure 2A | Figure 2B |
|---|---|
| Figure 2A. ALCL, low power magnification of hematoxylin and eosin stained skin sections Figure 2B. High power magnification of ALCL, “hallmark” cells with horseshoe-shaped nuclei are present. | |
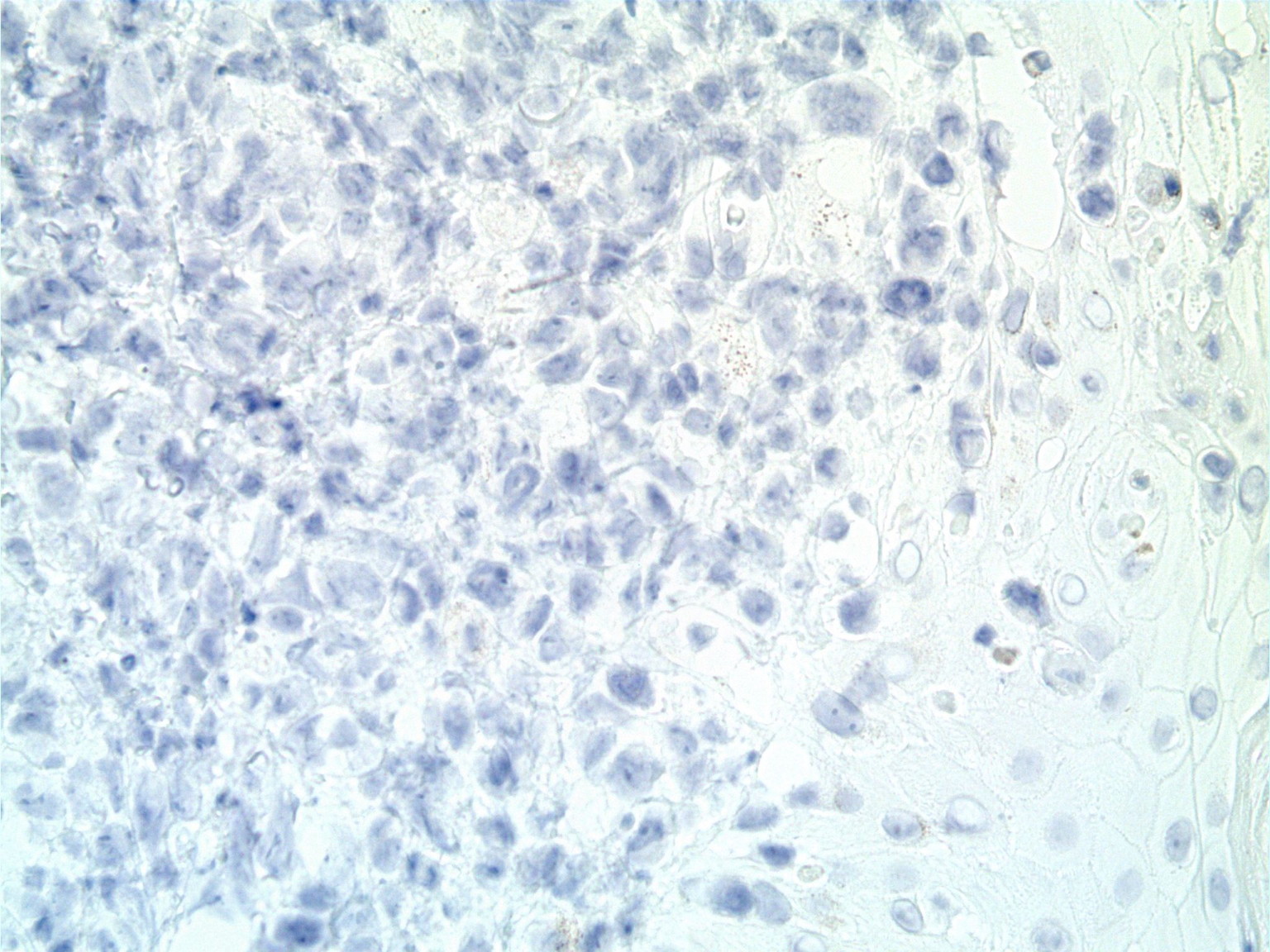
The atypical lymphocytes were characterized by “horseshoe” shaped nuclei and brightly eosinophilic perinuclear inclusions corresponding to the location of the Golgi apparatus (Figure 2B). These cells were CD2, CD3, CD7, CD8, CD30 (Figure 3A), and CD43 positive and CD4, CD5, ALK-1 (Figure 3B), CD34, CD117, myeloperoxidase, and CD20 negative.
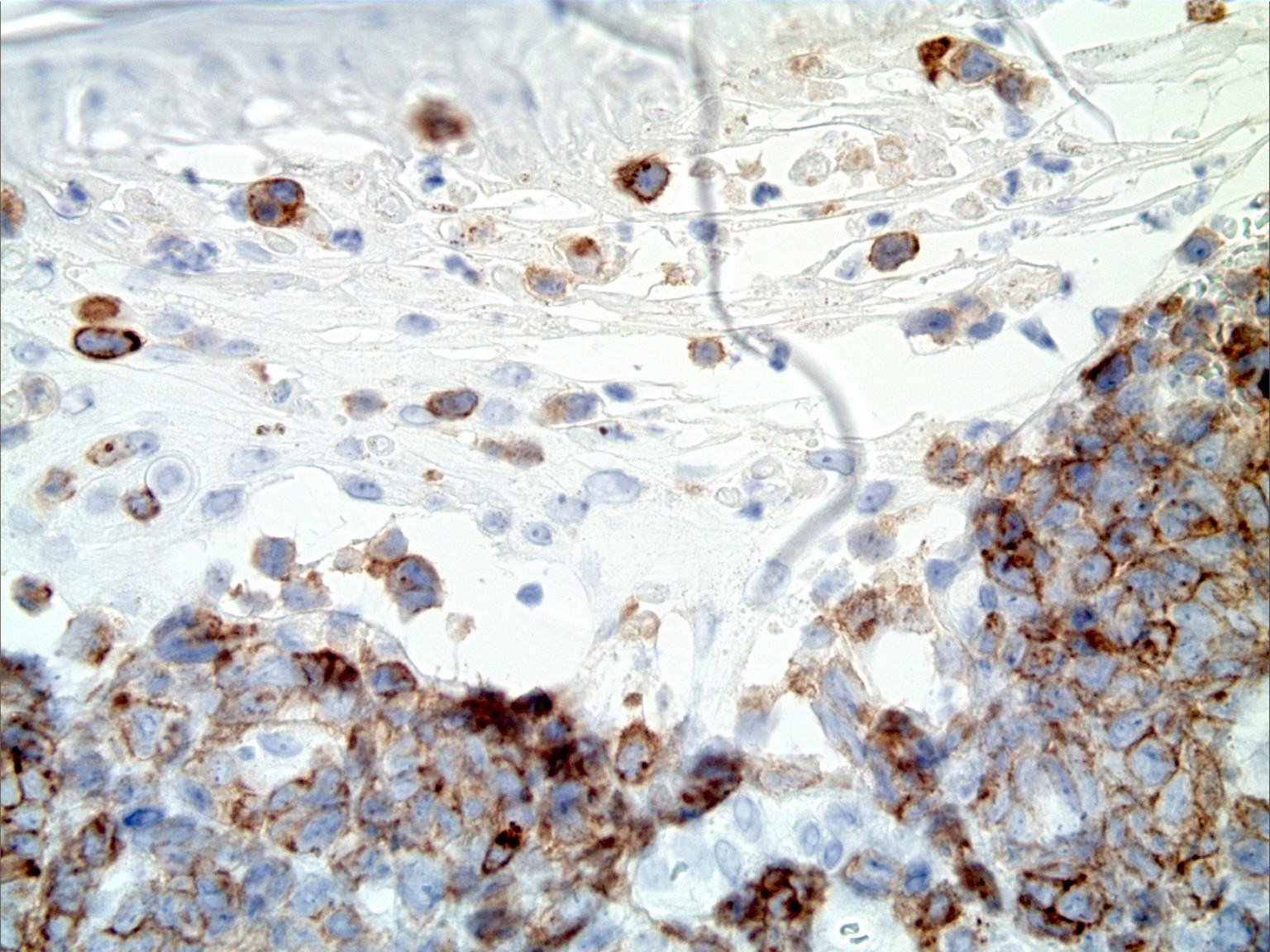 |  |
| Figure 3A | Figure 3B |
|---|---|
| Figure 3A. Positive immunohistochemistry for CD30 Figure 3B. Negative immunohistochemistry for ALK-1 | |
A bone marrow biopsy showed hypocellular marrow with scattered atypical cells that stained positive for CD2, CD3, CD30, CD8, CD43, and CD7, consistent with Anaplastic Large Cell Lymphoma (ALCL). The clinical presentation together with the skin and bone marrow biopsy results were diagnostic of ALK-negative S-ALCL. In addition, a cerebral spinal fluid specimen exhibited large atypical cells with a high nuclear-to-cytoplasmic ratio, markedly irregular nuclear contours, and vacuolated blue cytoplasm, consistent with involvement of the central nervous system. Furthermore, computed tomography of the chest, abdomen, and pelvis demonstrated hepatosplenomegaly and an enlarged pre-tracheal lymph node, consistent with widely metastatic disease. During the patient’s short hospital course, he became increasingly cachectic and delirious. He was started on salvage chemotherapy with cyclophosphamide and dexamethasone in an attempt to allow bone marrow recovery. Definitive chemotherapy was not started because his health deteriorated rapidly and he expired three weeks after admission.
Discussion
Systemic Anaplastic Large Cell Lymphoma (S-ALCL) was first described in 1985 as a subset of CD30-positive non-Hodgkin lymphomas [1]. In 2008, the World Health Organization (WHO) recognized two classifications of ALCL based on ALK-1 expression [2]. Currently, ALCL comprises approximately 3 percent of all non-Hodgkin lymphomas [3]. Extensive focus has been given to the ALK-positive ALCL group. However, the ALK-negative ALCL group remains poorly defined and lacks a consensus on histologic and immunohistochemical criteria for diagnosis [4-6]. Despite the ambiguity, the ALK-negative ALCL group comprises approximately 15-20 percent of all S-ALCL cases [7]. Compared to ALK-positive S-ALCL, ALK-negative S-ALCL is often seen in older age groups and is generally believed to portend a poor clinical course and outcome [5, 6]. The 5-year survival for ALK-negative S-ALCL ranges from 15-45 percent compared to 71-100 percent for ALK-positive S-ALCL [6, 8].
In addition to classification of S-ALCL by ALK positivity, the WHO also recognizes a variant of ALCL that is limited to the skin, termed primary cutaneous ALCL (cut-ALCL). Cut-ALCL is uniformly ALK-negative and is considered a non-Hodgkin lymphoma. Similar to ALK-negative S-ALCL, cut-ALCL tends to strongly express CD30 and the T cell markers CD2 and CD3 [9]. Microscopically, cutaneous lesions of cut-ALCL and ALK-negative S-ALCL both typically demonstrate dermal infiltrates of large lymphoid cells without epidermotropism [9]. As a result, histology alone cannot reliably distinguish between cut-ALCL and ALK-negative S-ALCL with cutaneous involvement. Complicating matters further, the clinical manifestations of cut-ALCL and ALK-negative S-ALCL with metastases to the skin can be strikingly similar. Both can exhibit erythematous, crusted papules and nodules that wax and wane [10]. Yang et al described the importance of systemic evaluation in cases of ALCL regardless of ALK-1 status because ALK-1 is not a reliable predictor for systemic involvement [11]. A thorough work-up to exclude systemic disease is prudent in all cases of ALCL presenting with skin lesions.
Whereas the histologic presentation of S-ALCL can vary from small cells to large anaplastic cells, almost all cases have classic “hallmark” cells present [12]. These are large cells with abundant cytoplasm, horseshoe-shaped nuclei, and a brightly eosinophilic region corresponding to the Golgi apparatus [12, 13]. Sometimes the nucleus completely surrounds this region. Histologically, S-ALCL also exhibits invasion of lymphatic vessels and a perivascular pattern of neoplastic cell infiltration [14]. Unfortunately, lesions of cut-ALCL have similar morphologic features.
Patients with S-ALCL typically present with painless lymphadenopathy and B symptoms (fever, night sweats, weight loss); 60 percent have advanced disease (stage III to IV) at presentation [6]. In contrast, cut-ALCL patients lack associated systemic findings at presentation. Polychemotherapy is the treatment of choice for both ALK-negative and ALK-positive S-ALCL patients. ALK expression independently predicts the survival difference among S-ALCL patients regardless of the chemotherapeutic regimen. Our patient exhibited the classic B symptoms of fever, weight loss, and lymphadenopathy. His disease was advanced at diagnosis and is similar to previously reported cases of rapidly progressing ALK-negative S-ALCL, except that most previously described patients did not demonstrate widespread skin disease.
Marshalko et al described a case of a 49-year-old male who was diagnosed with LyP during childhood, which was well-controlled throughout his life, who later developed a refractory case of ALK-negative S-ALCL with skin manifestations and tumor infiltration of the liver [15]. The patient died 20 months later of liver failure. Previous manifestations of his LyP/ALCL were treated successfully with cyclophosphamide, doxorubicin, oncovin, and prednisolone (CHOP) chemotherapy. However, his final flare was resistant to CHOP, hypercyclophosphamide, doxorubicin, vincristin dexamethasone (Hyper-CVAD), gemcitabine, cisplatin, methylprednisolone, thalidomide, and bortezomib therapy [15].
Autologous stem cell transplant in conjunction with high dose chemotherapy and radiation has been evaluated and resulted in long-term survival of 16 patients with chemotherapy-sensitive ALK-negative S-ALCL [16]. Thirteen of these 16 patients developed recurrent ALCL, of which 10 died. The median overall survival was 72 weeks [16]. Instead of long-term disease-free survival post-therapy, the majority of patients developed recurrent ALK-negative S-ALCL, which exhibited rapid progression and death of the patient. In the case of our patient, we were unable to assess the reactivity of his S-ALCL to chemotherapy and the case resulted in a similar outcome.
It is difficult to predict which cases of S-ALCL will be treatment responsive and which cases will be rapidly fatal. Factors that impair apoptosis negatively impact prognosis in S-ALCL [6]. An increase in cytotoxic T-lymphocytes (CTL) at ALCL tumor sites has been associated with a poor clinical outcome because CTLs select for apoptosis- and chemotherapy-resistant tumor cells, thus greatly increasing the clinical aggressiveness of the tumor [6]. Additionally, tumor cells that are deficient in caspase 3 (an apoptosis-inducing enzyme) and/or are actively expressing apoptosis-inhibiting proteins such as PI9 and Bcl-2 are correlated with a poor prognosis [6].
In summary, ALK-negative S-ALCL is a non-Hodgkin lymphoma that may exhibit cutaneous metastases at the time of presentation. The clinical and histologic features of cutaneous lesions are indistinguishable from those seen in cut-ALCL. Given the significant difference in prognosis between ALK-negative S-ALCL and cut-ALCL, a work-up for systemic involvement is needed in all patients that present with cutaneous lesions of ALCL in order to establish the correct diagnosis. Our case of rapidly fatal ALK-negative S-ALCL highlights the need for an improved diagnostic classification system that better predicts prognosis among patients at the time of presentation. Such advances in classification would hopefully translate into improved treatment strategies for this potentially fatal disease.
References
1. Mason DY, Harris NL, Delsol G. Anaplastic large cell lymphoma, ALK-negative. In: Swerdlow S. Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008. p. 317-9.2. Stein H, Mason DY, Gerdes J, O’Connor N, Wainscoat J, Pasllesen G, et al. The expression of the Hodgkin’s disease-associated antigen Ki-1 in reactive and neoplastic lymphoid tissue; evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985; 66:848-58. [PubMed]
3. Lu Y, Zhao X, Wang E, Chen W, Huang Q. ALK-negative anaplastic large cell lymphoma with extensive peripheral blood and boon marrow involvements manifested as “leukemic phase.” Leukemia Research. 2010; 34:475-82. [PubMed]
4. Ralfkier E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow H, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC) 2008:300-01.
5. Medeiros JL, Elenitoba-Johnson KSJ. Anaplastic Large Cell Lymphoma. Am J Clin Pathol. 2007; 127:707-22. [PubMed]
6. ten Berge, RL, Oudejans JJ, Ossenkoppele GJ, Meijer CJML. ALK-negative systemic anaplastic large cell lymphoma: differential diagnostic and prognostic aspects – a review. J Pathol. 2003; 200:4-15. [PubMed]
7. Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, Stroud D, et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal anti-body ALK1. Blood. 1997; 89:1394-404. [PubMed]
8. Gascoyne RD, Aoun P, Wu D, et al. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood. 1999; 93:3913-21. [PubMed]
9. Savage KJ, Harris NL, Vose JM, Ullrich F, et al. ALK-negative anaplastic large-cell lymphoma (ALCL) is clinically and immunophenotypically different from both ALK-positive ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008; 111:5496-5504. [PubMed]
10. L Cerroni. Lymphoproliferative lesions of the skin. J Clin Pathol. 2006 August; 59(8): 813-826. [PubMed]
11. Yang S, Wahlgren C, Ho J, Jukic D, Geskin L, and English JC. Cutaneous anaplastic large-cell lymphoma should be evaluated for systemic involvement regardless of ALK-1 status: case reports and review of literature. Am J Clin Dermatol. Jun 1; 12(3): 203-9. [PubMed]
12. Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, et al. ALK-Positive Lymphoma: A Single Disease With a Broad Spectrum of Morphology. Blood. Vol. 91 No. 6 (March 15), 1998: pp. 2076-2084. [PubMed]
13. ES Jaffe. Anaplastic large cell lymphoma: the shifting sands of diagnostic hematopathology. Mod Pathol. 2001; Mar 14(3):219-28. [PubMed]
14. Fornari, A, Piva R, Chiarle R, Novero D, Inghirami G. Anaplastic large cell lymphoma: one or more entities among T-cell lymphoma? Hematol Oncol. 2009; Dec 27(4):161-70. [PubMed]
15. Marschalko M, Eros N, Hollo P, Harsing J, et al. Secondary ALK Negative Anaplastic Large Cell Lymphoma in a Patient with Lymphomatoid Papulosis of 40 years Duration. Am J Dermatopathol. 2010 October; 32(7):708-712. [PubMed]
16. Zamkoff KW, Matulis MD, Mehta AC, Beaty MW, Hutchison RE, Gentile TC. High dose therapy and autologous stem cell transplant does not result in long-term disease-free survival in patients with recurrent chemotherapy-sensitive ALK-negative anaplastic large-cell lymphoma. Bone Marrow Transplant. 2004 Mar; 33(6):635-38. [PubMed]
© 2012 Dermatology Online Journal