Polyarteritis Nodosa
Published Web Location
https://doi.org/10.5070/D329p7j0pqMain Content
Polyarteritis Nodosa
Leonard H. Kim,MD
Dermatology Online Journal 7(1): 17
Department of Dermatology, New York UniversityPATIENT: 52-year-old woman
DURATION: Two months
DISTRIBUTION: Lower extremities
History
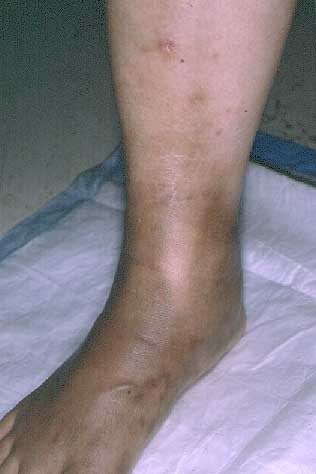
The patient presented to the Bellevue Hospital Medical Center Clinic in October, 1999 with a two-year history of pink, tender plaques on the lower extremities. An initial biopsy specimen at that time was interpreted as morphea, and she was treated with doxycycline 100 mg twice daily. After six weeks, her condition worsened with increased warmth and edema of the lower extremities. In early January, 2000, tender nodules erupted over the lower extremities. There were no systemic manifestations.
A biopsy specimen was repeated in January, 2000, and she was started on prednisone in the middle of February which produced improvement.
 |  |
| Figure 1 | Figure 2 |
|---|
Physical examination
Multiple, tender, erythematous nodules and hyperpigmented circumferential plaques were present over the lower extremities. Ulcers and livedo reticularis were not present.
Laboratory data
A urinalysis and complete blood count were normal. Erythrocyte sedimentation rate was 110 mm/hr, blood urea nitrogen 16 mg/dl, serum creatinine 0.7 mg/dl, aspartate aminotransferase 79 U/L, alanine aminotransferase 100 U/L, and alkaline phosphatase was 106 U/L. Hepatitis B surface antibody was positive. Hepatitis B surface antigen and hepatitis C antibody were negative. Antinuclear antibody, P-ANCA and C-ANCA were negative.
Histopathology
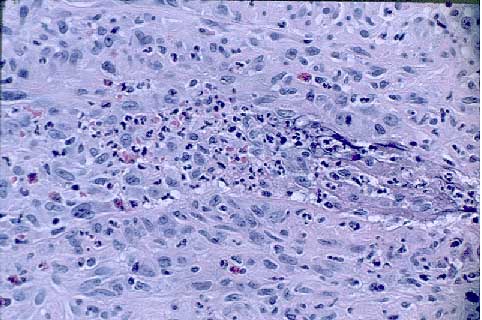
There were medium-sized blood vessels showing leukocytoclasis, fibrinoid necrosis, and transmural inflammation of their walls with a mixed inflammatory infiltrate containing neutrophils, lymphocytes, and some eosinophils.
Diagnosis
Polyarteritis nodosa
Comment
Classic polyarteritis nodosa is a systemic vasculitis that involves medium-sized muscular arteries in multiple organ systems.[1,2] The kidneys are the most frequently involved, followed by the liver, heart, gastrointestinal tract, musculoskeletal system, and central nervous system. Renal involvement can lead to glomerulonephritis and hypertension, which are leading causes of morbidity and mortality.
Approximately 25 percent of cases of classic polyarteritis nodosa will have cutaneous involvement. The most common cutaneous presentations include nodules, ulcers, and a livedo reticularis pattern on the lower extremities. The nodules are usually deep and painful, and may occur on the trunk and upper extremities.
Benign cutaneous polyarteritis nodosa is a disease that is limited to the skin, muscles, and joints.[3] The cutaneous lesions are indistinguishable from those of classic polyarteritis nodosa. Nodules are more common in the cutaneous variant. Four cases have been reported in which there was progression from cutaneous polyarteritis nodosa to systemic polyarteritis nodosa. In a study of 38 patients observed for up to 27 years with cutaneous polyarteritis nodosa, none developed systemic involvement.
The cause of both forms of polyarteritis nodosa is unknown, but an autoimmune etiology has been proposed. This is supported by direct immunofluorescence studies, which show immune complex deposits of IgM, C3, and fibrin. Polyarteritis nodosa has been associated with hepatitis B virus, hepatitis C virus, Crohn's disease, Takayasu's arteritis, relapsing polychronditis, streptococcal infections, and tuberculosis.[4]
Before the use of glucocorticoids, five-year survival of individuals with polyarteritis nodosa was ten percent if untreated. Today the five-year survival rate approximates 96 percent with the use of glucocorticoids and other immunosuppressive agents, such as cyclophosphamide. Patients with systemic involvement usually require high dose glucocorticoid therapy, frequently for years. Patients with primarily cutaneous involvement will often achieve remission within three to six months on lower doses of glucocorticoids. The general range of glucocorticoid dosages is one mg/kg/day of prednisone or its equivalent. Severe cases may be treated initially with methylprednisolone pulses. Cyclophosphamide is usually given at 50 to 100 mg/day. Sulfapyridine has been shown to help induce remission in the cutaneous variant.
Immunosuppressive agents like glucocorticoids and cyclophosphamide have been shown to perpetuate chronic hepatitis B virus infection and facilitate progression to cirrhosis. In hepatitis B virus-associated polyarteritis nodosa, glucocorticoids should be started initially to control the most severe life threatening manifestations of polyarteritis nodosa and then rapidly withdrawn. Concomitant antiviral agents such as viradibine, and interferon alfa-2b enhance survival.
References
1. Guillevin L, Lhote F. Treatment of polyarteritis nodosa and microscopic polyangiitis. Arthritis Rheum 1998;41(12):2100-5. PubMed2. Guillevin L. Treatment of classic polyarteritis nodosa in 1999 [editorial] Nephrol Dial Transplant 1999;14(9):2077-9. PubMed
3. Pak H, Montemarano AD, Berger T. Purpuric nodules and macules on the extremities of a young woman. Cutaneous polyarteritis nodosa. Arch Dermatol 1998;134(2):231-2, 234-5. PubMed
4. Soufir N, Descamps V, Crickx B, Thibault V, Cosnes A, Becherel PA, Wolkenstein P, Bournerias I, De La Salmoniere P, Picard C, Roujeau JC, Piette JC, Belaich S, Revuz J, Frances C. Hepatitis C virus infection in cutaneous polyarteritis nodosa: a retrospective study of 16 cases [letter] Arch Dermatol 1999;135(8):1001-2. PubMed
© 2001 Dermatology Online Journal