Dyschromatosis universalis hereditaria: Two cases
Published Web Location
https://doi.org/10.5070/D329m212t0Main Content
Dyschromatosis universalis hereditaria: Two cases
N Kenani 1, N Ghariani 1, M Denguezli 1, B Sriha2, C Belajouza 1, R Nouira 1
Dermatology Online Journal 14 (2): 16
1. Department of Dermatology, Chu Farhat Hached Sousse, Tunisia 2. Department of Pathology, Chu Farhat Hached Sousse, Tunisia
Abstract
Dyschromatosis universalis hereditaria is a rare genodermatosis reported initially and mainly in Japan. However, subsequent cases have been reported from other countries. We describe two Tunisian cases of dyschromatosis universalis hereditaria in a 3-year-old and a 3-month-old girl. They presented to our department with asymptomatic progressive mottled pigmentation over the trunk and limbs, which had been noted since birth and had become more noticeable with age. Palms and soles were also affected in the first case. The two patients did not have any systemic or other cutaneous illness. They were born to healthy, second-degree consanguineous parents (case 1) and non consanguineous parents (case 2), following an uneventful pregnancy. No family members had a similar appearance. Physical examination revealed numerous, generalized, hyperpigmented macules interspersed with spotty de-pigmented macules. Hair, nails, teeth, and mucosae were normal. Systemic examination did not reveal abnormalities. Histological exam revealed basal layer hypermelanosis with pigmentary incontinence in some areas. So based on those findings a clinical diagnosis of DUH was made and the patients were followed up in our department for periodic general evaluation of their skin. After a follow up of 12 months, the first child didn't develop other lesions, but palms and soles were also involved in the second case.
Dyschromatosis universalis hereditaria (DUH) is an extremely rare genodermatosis, characterized by hyper- and hypo-pigmented macules forming a reticulate pattern. The initial case and most reported cases are in Japanese literature. However, subsequent cases have been reported from other countries. We describe two Tunisian girls with features of DUH whose family members were not affected.
Case 1
 |  |
| Figure 1 | Figure 2 |
|---|
 |  |
| Figure 3 | Figure 4 |
|---|---|
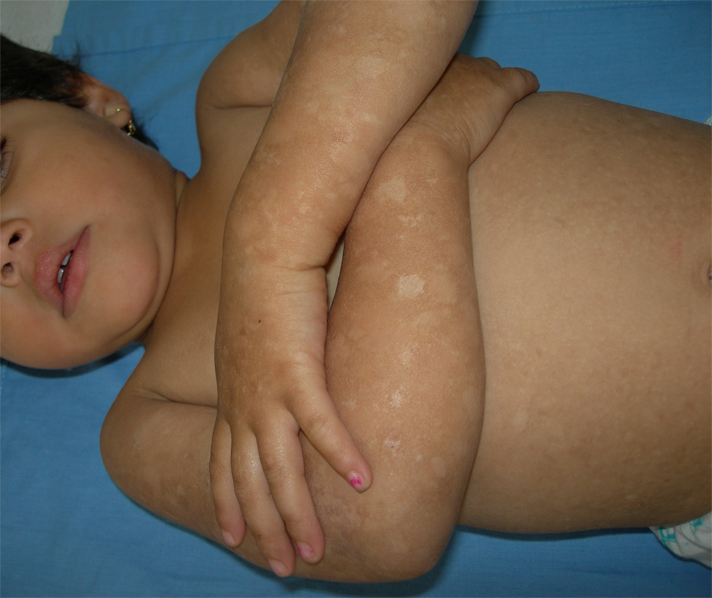
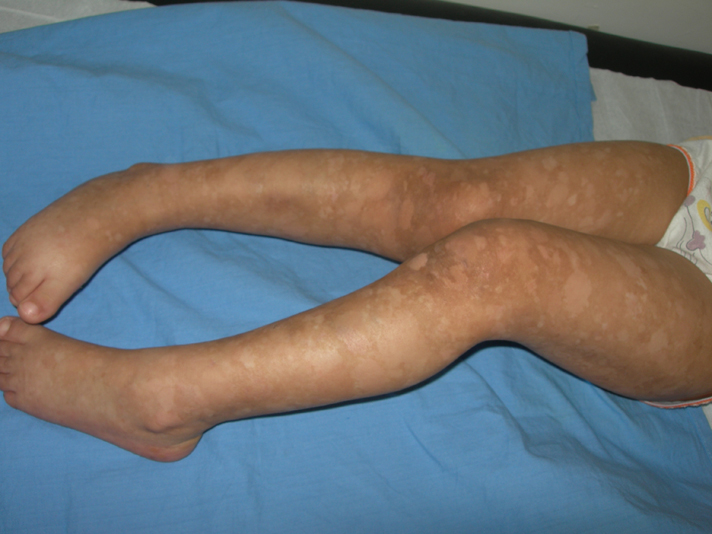
| Figures 1-3. (case 1) : Hyperpigmented macules interspersed with spotty hypopigmented macules denser on the limbs than on
the trunk Figure 4. (case 1) involvement of soles | |
 |
| Figure 5 |
|---|
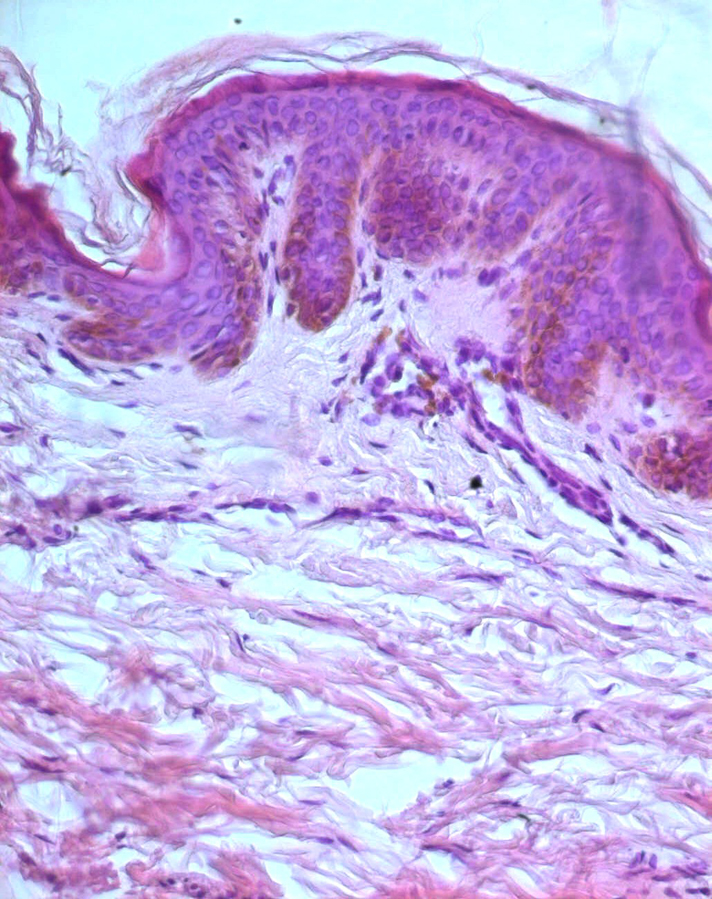
| Figure 5. (case 1) HE X100: Basal layer hypermelanosis and pigment incontinence in some areas of the upper dermis |
A 3-year-old girl presented to our dermatology department with asymptomatic progressive mottled pigmentation over the trunk and limbs, which had been noted since birth and had become more noticeable with age. She did not have any systemic or other cutaneous illness. There was no history of photophobia or photosensitivity. The girl was born to healthy, second-degree consanguineous parents, following an uneventful pregnancy. No family members had a similar appearance. Her mental and developmental milestones were normal. Physical examination revealed numerous asymptomatic, generalized, 0.5- 5 cm hyperpigmented macules interspersed with spotty hypopigmented macules. The lesions were denser on the limbs than on the trunk (Figs 1-3). Palms and soles were also involved (Fig. 4). Her face was not involved. The hair, nails, teeth, and mucosae appeared normal. There was no apparent atrophy, erythema, or telangiectasia. Systemic examination revealed no abnormalities. Biopsy from the hyperpigmented lesions on the back showed basal layer hypermelanosis and pigment incontinence in some areas of the upper dermis (Fig. 5). Based on those findings a clinical diagnosis of DUH was made and the patient was followed up in our department for periodic general evaluation of their skin. After a follow up of 12 months, skin lesions were stabilized.
Case 2
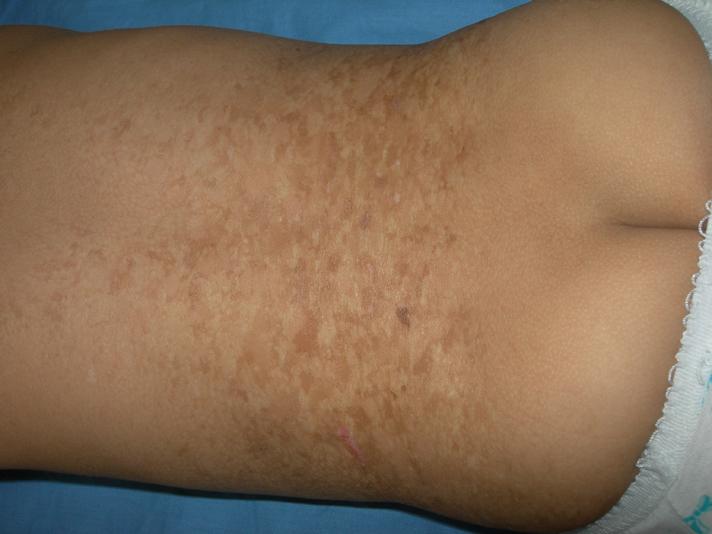
A 3-month-old female infant, born of non-consanguineous parents after a normal full term vaginal delivery, presented to the dermatology department for generalized hyperpigmented and hypopigmented macules that had been noted since birth. Her developmental milestones were normal. She did not have any other affection. There was no history of a similar disorder in her family. The clinical examination revealed generalized, well-demarcated, 0.5- 5 cm hyperpigmented and hypopigmented macules, predominating on limbs (Figs. 6-9). The trunk was also involved, but the face, palms and soles were spared. The hair, nails, teeth, and mucosae were normal. Systemic examination was normal. Histological examination was similar to the first case. These findings were consistent with the diagnosis of DUH. The patient was regularly followed up in our department. Skin lesions had progressive involvement and affected palms and soles after a followup of 12 months.
 | 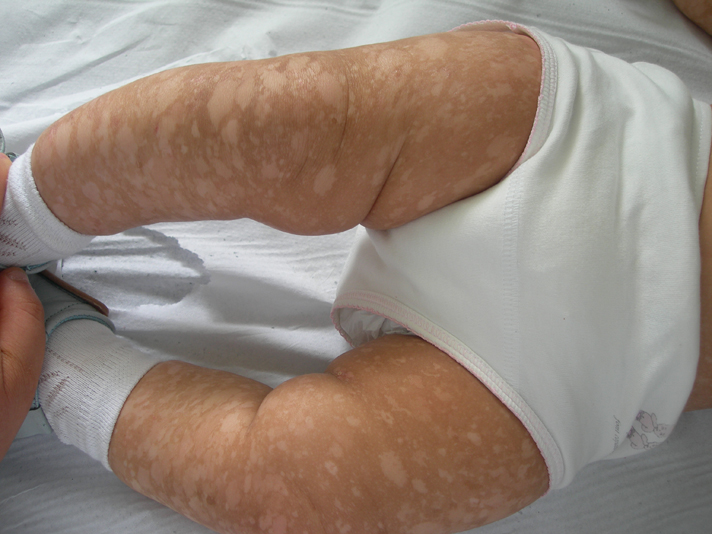 |
| Figure 6 | Figure 7 |
|---|
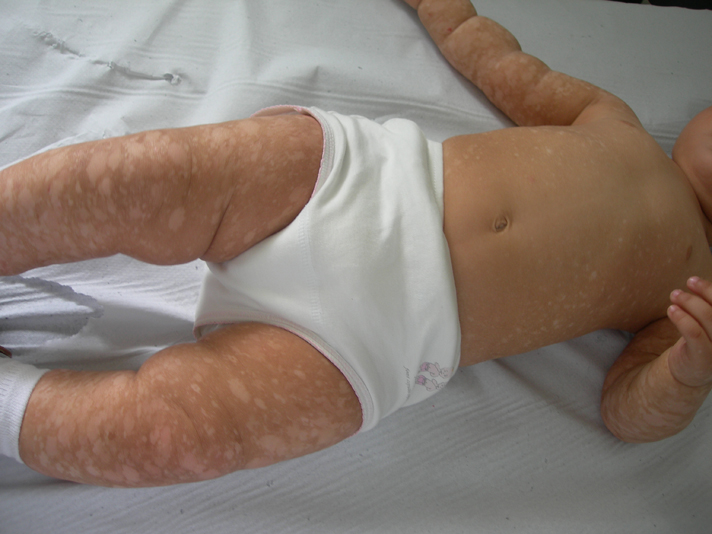 |  |
| Figure 8 | Figure 9 |
|---|---|
| Figures 6-9 (case 2) Generalized, well demarcated, hyperpigmented and hypopigmented macules, predominating on limbs and trunk and sparing the face | |
Discussion
Dyschromatoses are a group of disorders characterized by the presence of small and irregular shape hyper and hypopigmented macules. Two major types have been described, based on distribution of lesions as follows: dyschromatosis universalis hereditaria (DUH), first reported in 1933 by Ichikawa and Higara [1], in which mixtures of hyper and hypopigmented macules occur all over the body; and dyschromatosis symmetrica hereditaria (DSH), also known as acropigmentation of Dohi, described in 1929 by Toyama [2], which is a localized form involving only the acral areas. Both conditions are seen most commonly in Japan. However, a few cases of DUH were described among Europeans, South Americans, Indians, and Saudi Arabians [3, 4, 5]. Tunisian cases of DUH are exceptional, only two percent cases were reported, in two younger men with a family history in one case [6].
The etiology of this disorder is not yet known. A variable autosomal mode of inheritance of DUH has been described. No family members had a similar appearance in our two cases. Some clinicians have suggested that DSH might be a subtype of DUH. The RNA-specific adenosine deaminase gene (ADAR1, DSRAD) responsible for DSH was firstly described in Japanese families [7]. After that, novel mutations in the ADRA1 were reported in Chinese families confirming that this gene is responsible for DSH in different ethnic group [8, 9]. More recently, Suzuki et al. reported 16 novel mutations in the ADRA1 without identifying such as mutations in patients with DUH [10]. Furthermore, they did not find the two diseases together in the same pedigree indicating that these two conditions are completely different.
In DUH, skin lesions are usually present in the first years of life. The trunk and extremities are the dominant sites. Facial lesions were seen in almost 50 percent of affected individuals but involvement of palms and soles is unusual [5]. Abnormalities of hair and nails have also been reported [3].
In our two cases, faces were spared but palms and soles were involved. The hair, nails, teeth, and mucosae appeared normal. In comparison with the literature, the precocity of skin involvement in our two younger patients is noticeable, mainly in the second case.
The histopathology typically shows a focal increase or decrease in melanin content of the basal layer (depending on the type of the lesion biopsied) and occasionally pigmentary incontinence. In a recent ultrastructural skin investigation Nuber et al. indicated that DUH is a disorder of melanosome synthesis rate or in melanocyte activity and not a disorder of melanocyte number [11].
Dyschromatosis universalis hereditaria may be associated with abnormalities of dermal connective tissue, nerve tissue, or be associated with other systemic complications [4, 5]. No such features characterized our patients.
Many diseases characterized by cutaneous dyschromia must be differentiated from DUH. The most important one in our country is xeroderma pigmentosum (XP) [4, 5, 6], which is more frequent. Our patients had no signs of XP (no poikiloderma, atrophy, or telangiectasia).
Generally, DUH does not progress or worsen with age. In all reports there was no spontaneous regression.
References
1. Ichikawa T, Higara Y. A propos d'une anomalie pigmentaire inédite. Jpn J Dermatol 1933 ; 34 : 360-4.2. Toyama I. Dyschromatosis symmetrica herediteria. Jpn J Dermatol 1929 ; 29 : 95-6.
3. Sethuraman G, D'Souza M, Mohan Thappa D, Srinivas CR, Smilest L. Clin Exp Dermatol 2002 ; 27 : 477-479.
4. Bukhari IA, El-Harith EA, Stuhrmann M. Dyschromatosis universalis hereditaria as an autosomal recessive disease in five members of one family. J Eur Acad Dermatol Venereol. 2006; 20(5): 628-9.
5. Al Hawsawi K, Al Aboud K, Ramesh V, Al Aboud D. Dyschromatosis universalis hereditaria: report of a case and review of the literature. Pediatr Dermatol. 2002; 19: 523-526.
6. Dhaoui MA, Doss N. Dychromatose universelle: 2 cas. Ann Dermatol Venereol. 2001; 128: 136-8.
7. Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, Tomita Y: Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet 2003; 73:693-699.
8. Li M, Jiang YX, Liu JB, et al: A novel mutation of DSRAD gene in a Chinese family with dyschromatosis symmetrica hereditaria. Clin Exp Dermatol 2004; 29:533-535.
9. Liu Q, Liu W, Jiang L, et al: Novel mutations of the RNA-specific adenosine deaminase gene (DSRAD) in Chinese families with dyschromatosis symmetrica hereditaria. J Invest Dermatol. 2004;122:896-899.
10. Suzuki N, Suzuki T, Inagaki K, Ito S, Kono M, Fukai K, Takama H, Sato K, Ishikawa O, Abe M, Shimizu H, Kawai M, Horikawa T, Yoshida K, Matsumoto K, Terui T, Tsujioka K, Tomita Y. Mutation analysis of the ADAR1 Gene in Dyschromatosis Symmetrica Hereditaria and genetic differentiation from both Dyschromatosis Universalis Hereditaria and Acropigmentatio Reticularis. J Invest Dermatol. 2005; 124 : 1186-92.
11. Nuber UA, Tinschert S, Mundlos S, Hauber I. Dyschromatosis universalis hereditaria: familial case and ultrastructural skin investigation. Am J Med Gen A. 2004; 125(3): 261-6.
© 2008 Dermatology Online Journal