Denuded congenital lesions: Recessive dystrophic epidermolysis bullosa
Published Web Location
https://doi.org/10.5070/D32694z1t6Main Content
Denuded congenital lesions: Recessive dystrophic epidermolysis bullosa
Kristy F Fleming MD1, Jashin J Wu MD2, Senait W Dyson MD3, Soheil S Dadras MD PhD4, Brandie J Metz MD3
Dermatology Online Journal 15 (4): 4
1. Department of Internal Medicine, Kaiser Permanente Los Angeles Medical Center, Los Angeles, California2. Department of Dermatology, Kaiser Permanente Los Angeles Medical Center, Los Angeles, California
3. Department of Dermatology, University of California, Irvine, Irvine, California
4. Departments of Pathology and Dermatology, Stanford University, Stanford, California. bmetz@uci.edu
Abstract
Recessive dystrophic epidermolysis bullosa (Hallopeau-Siemens type) (RDEB-HS) is a rare severe mechanobullous disorder resulting from a defect in collagen VII. Patients with RDEB-HS present with generalized blistering and denudation of the skin at birth and have mucosal involvement. The repeated blistering leads to scarring, which may be deforming and result in serious complications. Transmission electron microscopy is currently the gold standard for diagnosis of RDEB-HS.
Case description
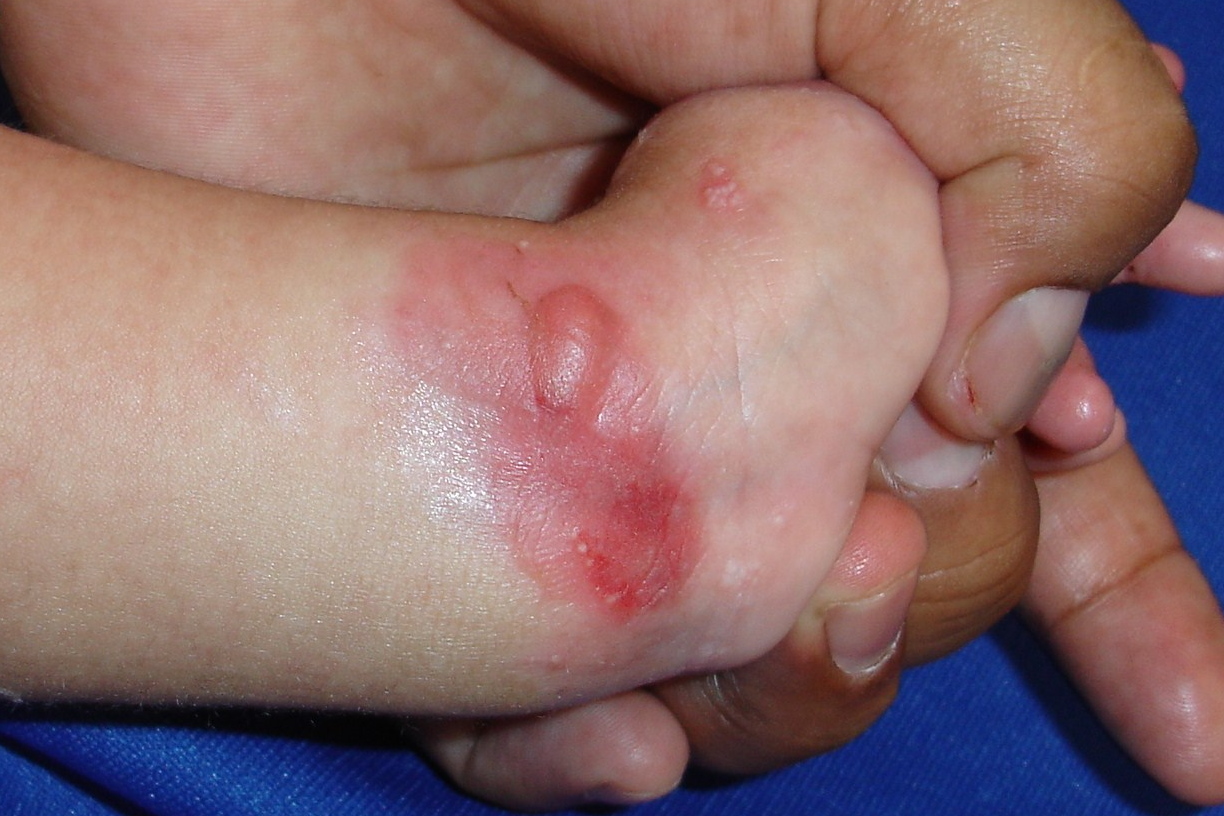 | 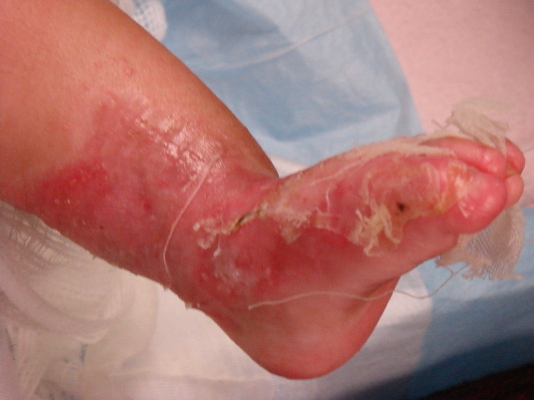 |
| Figure 1 | Figure 2 |
|---|
An infant girl presented on the third day of life with multiple areas of denuded skin. At birth she was noted to have denuded skin lesions involving both lower extremities and the left wrist (Figs. 1 & 2). She had since developed circumscribed lesions and blisters involving the right upper extremity, index finger, back, and left perianal region as well as bleeding blisters with whitish discoloration on the buccal mucosa and gums. She was the product of an uncomplicated term pregnancy and there was no family history of congenital malformation or blistering diseases.
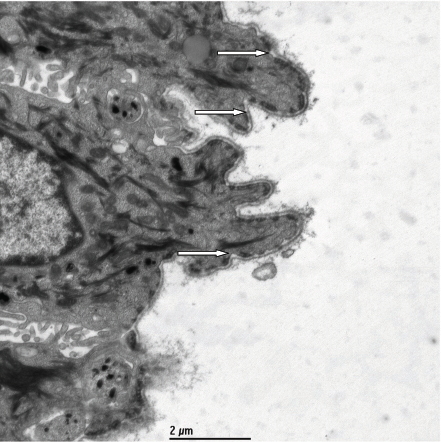 |
| Figure 3 |
|---|
In addition to the cutaneous findings, physical exam revealed hypertelorism, macrocephaly, mild retronathia, high-arched palate, and bilateral peri-auricular skin tags. The rest of the exam was unremarkable. A cutaneous biopsy and electron microscopy was performed (Fig. 3).
Immunohistochemical findings showed skin with a subepidermal cleft. Basement membrane associated antibodies to GB3 (laminin 5), 123 (LAD-1, BP180), and collagen IV were present in the roof of the blister of the patient's biopsy and equal in intensity to the normal control. Collagen VII was found in the normal skin control. However, collagen VII was absent in the patient's tissues, a finding consistent with recessive dystrophic epidermolysis bullosa.
Electron microscopy revealed that the roof of the blister was composed of intact basal keratinocytes, anchoring filaments, and an intact lamina densa. The skin division occurred below the level of the lamina densa. Anchoring fibrils within the dermis also appeared reduced. These findings are also consistent with recessive dystrophic epidermolysis bullosa. The patient was diagnosed with Recessive Dystrophic Epidermolysis Bullosa (Hallopeau-Siemens type) (RDEB-HS).
Discussion
Inherited epidermolysis bullosa (EB) is classified into three major types depending on the ultrastructural level where blisters develop, determined by electron microscopy: epidermolytic (EB simplex) at the level of the basal layer, lucidolytic (juntional EB) at the lamina lucida, and dermolytic (dystrophic EB) at the sub-lamina densa [1]. Each major type is further classified into subtypes and subcategories. Dystrophic EB is divided into dominant and recessive forms, with recessive dystrophic EB (RDEB) further classified into three categories: RDEB, severe generalized (also known as Hallopeau-Siemens type); RDEB, generalized other; and RDEB-bullous dermolysis of the newborn (RDEB-BDN). Recessive Dystrophic Epidermolysis Bullosa (Hallopeau-Siemens type) (RDEB-HS) is distinguished from Dominant DEB and the other sub-categories of RDEB by its characteristic ultrastructural findings and alterations in antigenic staining demonstrated by immunofluorescence mapping, as shown in Table 1, as well as its myriad of cutaneous and extracutaneous clinical manifestations as displayed in Table 2.
Recessive Dystrophic Epidermolysis Bullosa (Hallopeau-Siemens type) (RDEB-HS) is a severe inherited mechanobullous disorder characterized by tense blisters that arise in response to mild mechanical disruption. These friction-induced blisters result from a mutation in the collagen type VII gene that leads to dysfunctional anchoring fibrils, which are unable to form an interconnecting network across the dermal-epidermal basement membrane [2].
The most common genetic abnormality in RDEB-HS is the presence of a premature stop codon in both alleles of the COL7A1 gene, leading to the production of a truncated form of the type VII collagen protein [2]. The truncated protein formed is quickly degraded, explaining the negative immunohistochemical staining for type VII collagen [3].
The diagnosis of epidermolysis bullosa (EB) usually requires focused lab testing, such as transmission electron microscopy, immunofluorescence antigen mapping, or immunohistochemical staining with EB-specific monoclonal antibodies [4]. Transmission electron microscopy is the gold standard for diagnosing EB [4].
Most patients with dystrophic epidermolysis bullosa have the dominantly-inherited form of the disease, which presents in infancy or early childhood with acral blistering that heals with scarring, milia formation, and nail dystrophy [4]. The Hallopeau-Siemens variant of EB is a much more severe, mutilating disease. Recessive Dystrophic Epidermolysis Bullosa (Hallopeau-Siemens type) (RDEB-HS) presents with generalized blistering and denudation of the skin at birth, with mucosal involvement and pariodontitis along with enamel defects leading to dental caries [4]. Repeated blistering results in significant scarring that has far reaching sequelae. The systemic clinical features typical of RDEB-HS are consequences of the distortion of tissue by scarring and include: dysphagia and esophageal strictures leading to malnutrition, flexion contractures of the limbs, pseudosyndactyly, which may eventually lead to mitten deformity of the hands, microstomia, ankyloglossia, corneal surface scarring, urethral stenosis, and constipation secondary to anal fissure formation [2]. Patients with RDEB-HS are also at risk for developing potentially lethal squamous cell carcinoma in chronic recurrent erosions of the skin and mucosal membranes [5]. There is currently no satisfactory treatment available for RDEB-HS and the long-term prognosis is poor for this unremitting, debilitating disease. Management consists of continued wound care, infection control, nutritional support, and tailored therapies depending on the specific complications that arise.
References
1. Fine JD, Eady RAJ, Bauer EA et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931-50. [PubMed]2. Uttio J, Richard G. Progress in epidermolysis bullosa: from eponyms to molecular genetic classification. Clin Dermatol. 2005;23(1):33-40. [PubMed]
3. Cui Y, Hagan KW, Zhang S et al. Identification and characterization of genes that are required for the accelerated degradation of mRNAs containing a premature termination codon. Genes Dev. 1995;9:423-436. [PubMed]
4. Das BB, Sahoo S. Dystrophic epidermolysis bullosa. J Perinatol. 2004;24,41-47. [PubMed]
5. Horn HM, Tidman MJ. The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol. 2002;146:267-274. [PubMed]
© 2009 Dermatology Online Journal