Recessive dystrophic epidermolysis bullosa: Presentation of two forms
Published Web Location
https://doi.org/10.5070/D32348f29cMain Content
Recessive dystrophic epidermolysis bullosa: Presentation of two forms
Ljiljana Medenica1, Marko Lens2
Dermatology Online Journal 14 (3): 2
1. Department of Dermatovenereology, School of Medicine, University of Belgrade, Institute of Dermatovenereology, Clinical
Center of Serbia, Pasterova 2, Belgrade, Serbia. limed@EUnet.yu2. King's College, Genetic Epidemiology Unit, St.Thomas' Hospital, Lambeth Palace Road, London SE1 7EH, United Kingdom
Abstract
Dystrophic epidermolysis bullosa (DEB) and aplasia cutis congenita (ACC), also known as congenital localized absence of skin (CLAS) are rare clinical entities. Aplasia cutis congenita presented in conjunction with simplex, junctional, or dystrophic types of epidermolysis bullosa (EB) is classified as type-6 ACC. This association was initially described and referred in the literature as Bart syndrome. We describe two cases of recessive DEB (RDEB), one with the major Hallopeau-Siemens (RDEB-HS) subtype and one case with the minor RDEB inversa (RDEB-I) subtype associated with ACC localized on the lower extremities. Full clinical history and transmission electron microscopic findings are presented for both cases. To date, only five cases of RDEB presenting with ACC have been reported in the literature. Detailed descriptions of the association of RDEB and ACC in the literature are scarce. It seems that this condition is probably more common in clinical practice than described in the literature. Our findings confirm that the term, Bart syndrome, should not be considered as a separate entity or clinical variant of dominant dystrophic EB as it was initially described. Congenital localized absence of skin may be associated with any of the three major types of EB (simplex, junctional, or dystrophic).
Introduction
Epidermolysis bullosa (EB) is a heterogeneous group of mechanobullous, genetically determined disorders characterized by increased skin fragility and blistering of the skin following minor or insignificant trauma or traction of the skin [1]. A revised classification system for inherited EB recommended by an international committee of experts, based on the ultrastructural level of skin cleavage, clinical phenotype, and genotype designates three major EB types, simplex (epidermolytic EB), junctional, and dystrophic (dermolytic EB) with ten distinctive clinical subtypes [2]. Dystrophic EB (DEB) includes three major subtypes as follows: dominant DEB (DEB), recessive DEB Hallopeau-Siemens (RDEB-HS), and RDEB non-Hallopeau-Siemens (RDEB-nHS); and two minor subtypes as follows: RDEB inversa (RDEB-I) and RDEB centripetalis (RDEB-ce) [2].
Aplasia cutis congenita (ACC), also known as epitheliogenesis imperfecta or congenital absence of skin, is a rare clinical condition in which localized or widespread areas of skin are absent at birth [3]. According to the number and location of the skin defects and the presence or absence of associated malformations, Freiden proposed a classification of ACC identifying nine different types of ACC [4]. Aplasia cutis congenita presented in conjunction with simplex, junctional, or dystrophic types of EB is classified as type 6 ACC [4, 5]. This association was initially described and referred to in the literature as Bart syndrome [6, 7]. Since then, additional cases of simplex, junctional, dominant or recessive dystrophic EB in association with congenital localized absence of skin have been reported [8-13].
Recessive DEB associated with ACC (ACC type 6), presented as congenital absence of skin localized on the lower extremities is a very rare condition and currently only five patients with this association have been described in the published literature [5, 12]. We describe two additional cases of RDEB, one with the major Hallopeau-Siemens (RDEB-HS) subtype and one case with the minor RDEB inversa (RDEB-I) subtype associated with ACC localized on the lower extremities.
Clinical synopsis
Case 1.—A 27-year-old Caucasian man was referred to our Institute (Academic, Teaching Institute of Dermatovenereology, Clinical Center of Serbia) at his birth because of denuded skin on the lower legs and both feet and generalized blistering of the skin. The patient was examined and observed at our Institute on a regular basis since his birth.
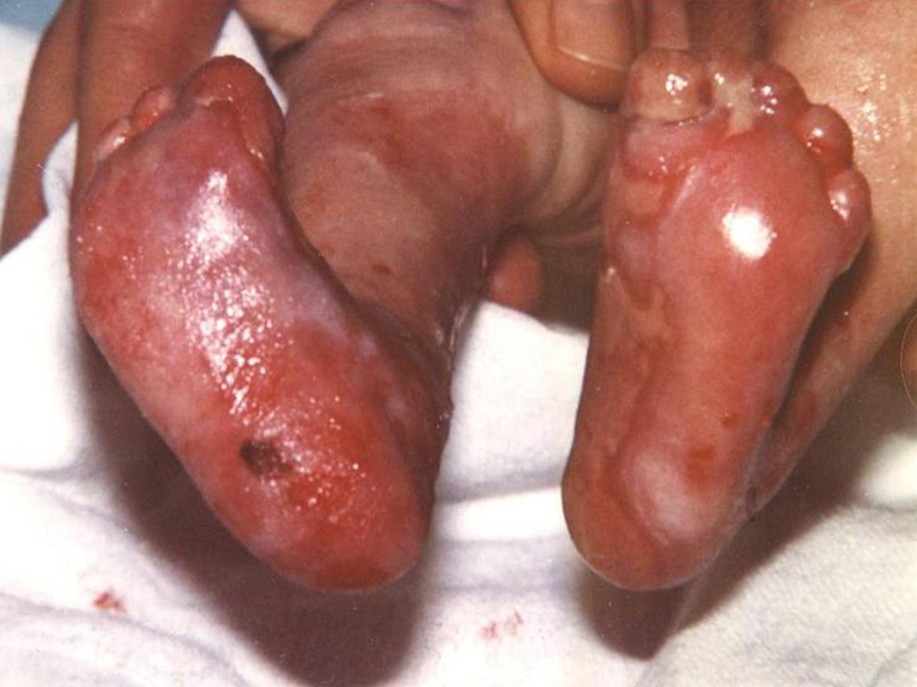
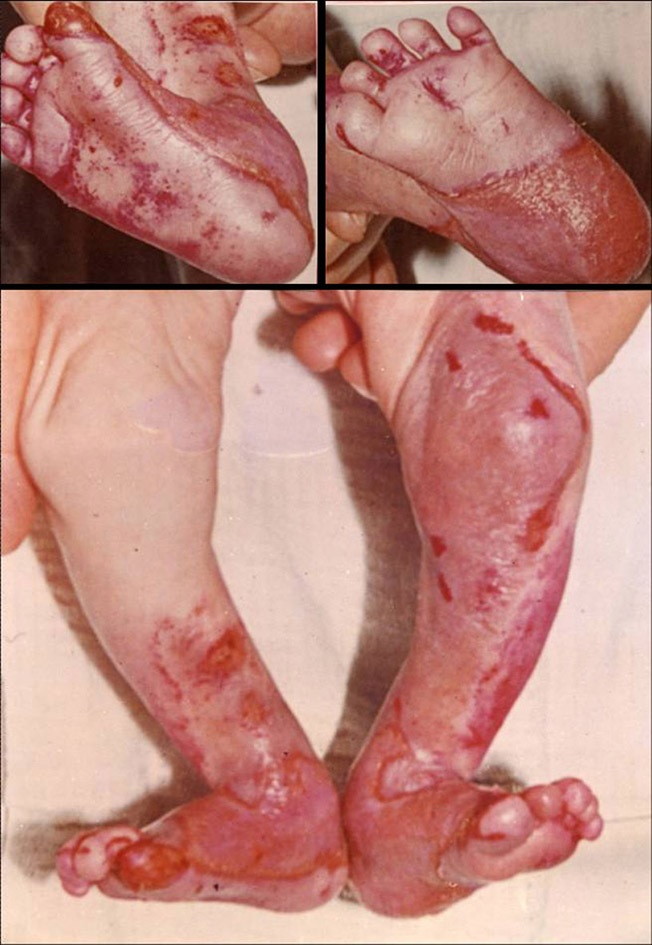
At birth the patient had bilateral skin defects with sharply demarcated borders covered by red, thin, moist glistening epithelial membrane on the anterior, medial and lateral aspect of the lower third of the right and left legs and feet, including plantar and dorsal sides of the toes (Fig. 1). The eroded areas healed within 1 month. Bilateral dorsiflexion of the feet, more pronounced on the right foot, was observed.
 |  |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. Congenital localized absence of the skin in patient with RDEB-HS: A. Glistening epithelial membrane observed on
the anterior, medial and lateral aspect of the lower third of the right and left legs and feet including plantar and dorsal
side of the toes; bilateral dorsal flexion of the feet, B. Pronounced dorsal flexion on the right foot. Figure 2. "Mitten-like" appearance of the left hand and feet (pseudosyndactyly) in the patient with RDEB-HS. | |
The patient had continuous development of blisters and erosions on the skin since birth. The blisters and erosions mainly developed on trauma-prone skin or elsewhere after minor trauma; some occurred spontaneously. Blisters tended to be localized predominantly on his hands, feet, elbows, knees and oral mucosa. All blisters healed with fine atrophic scars and milia formation. The fingers and toes were fused by scar tissue leading to a mitten-like appearance of hands and feet (pseudosyndactyly) (Fig. 2). Fingernails and toenails were absent from early childhood. Marked growth retardation and other severe extracutaneous complications developed. Ocular involvement has included symblepharon and corneal scaring. Mucous membranes have been highly involved since birth. The patient gradually became unable to protrude his tongue beyond the teeth. The teeth were highly carious or absent. Esophageal involvement characterized by dysphagia and pain on swallowing has been detected. Esophageal stenosis and spasm were diagnosed by radioscopy. The patient suffered also from severe, persistent iron deficiency anemia. Eroded skin was rarely free of secondary bacterial infection. There was no family history of EB or ACC.
 |  |
| Figure 3a | Figure 3b |
|---|---|
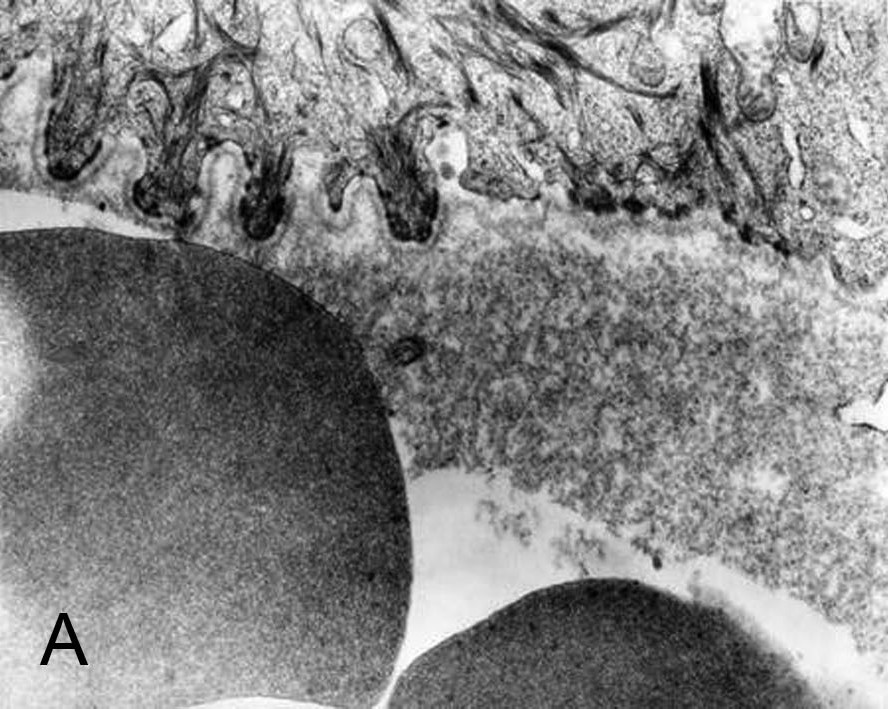
| Figure 3a. Electron micrograph showing blister formation formed below the basement membrane zone, just beneath the level of
lamina densa, and absent anchoring fibrils along the epidermal roof of the blister. Blister cavity contains erythrocytes,
collagen remnants and amorphous substance (36,000X). Figure 3b. Intact perilesional skin with complete absence of anchoring fibrils (97,000X). | |
A biopsy tissue specimen from perilesional intact skin and the edge of fresh blisters was obtained, cut into small fragments, and processed for transmission electron microscopy. Sublamina densa cleavage (dermolytic mode of blistering) was seen. In the blister roof total absence of anchoring fibrils was noticed; epidermal keratinocytes and hemidesmosomes were normal. The blister cavity contained erythrocytes, collagen, and amorphous substance (Fig. 3a). In the intact perilesional skin anchoring fibrils were also absent. In the blister floor, evidence of collagenolysis was pronounced; individual collagen fibrils had irregular diameters or a ghost-like appearance. In some areas, fine granular or amorphous deposits were found between the collagen fibrils, isolated or in bundles (Fig. 3b).
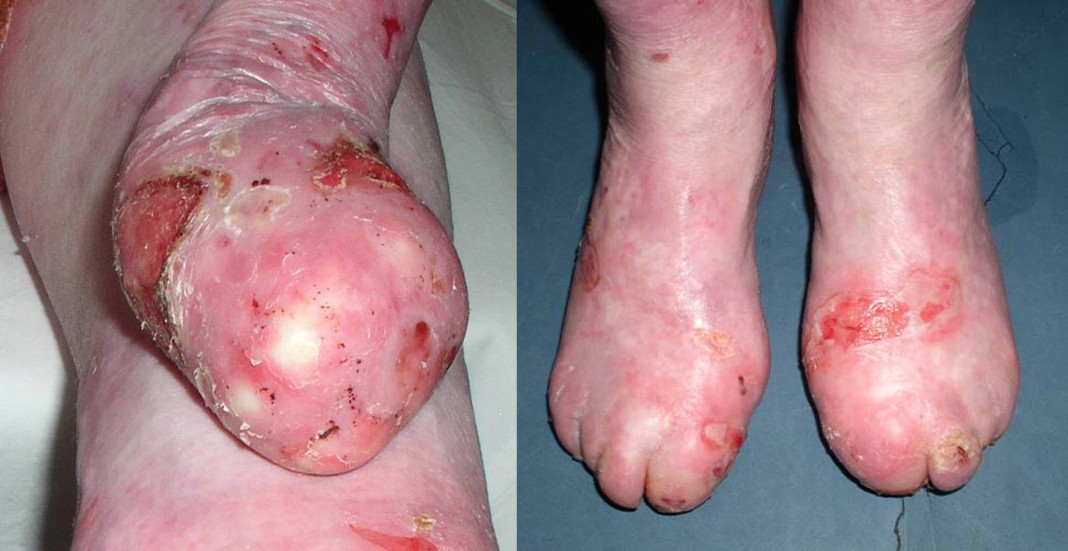
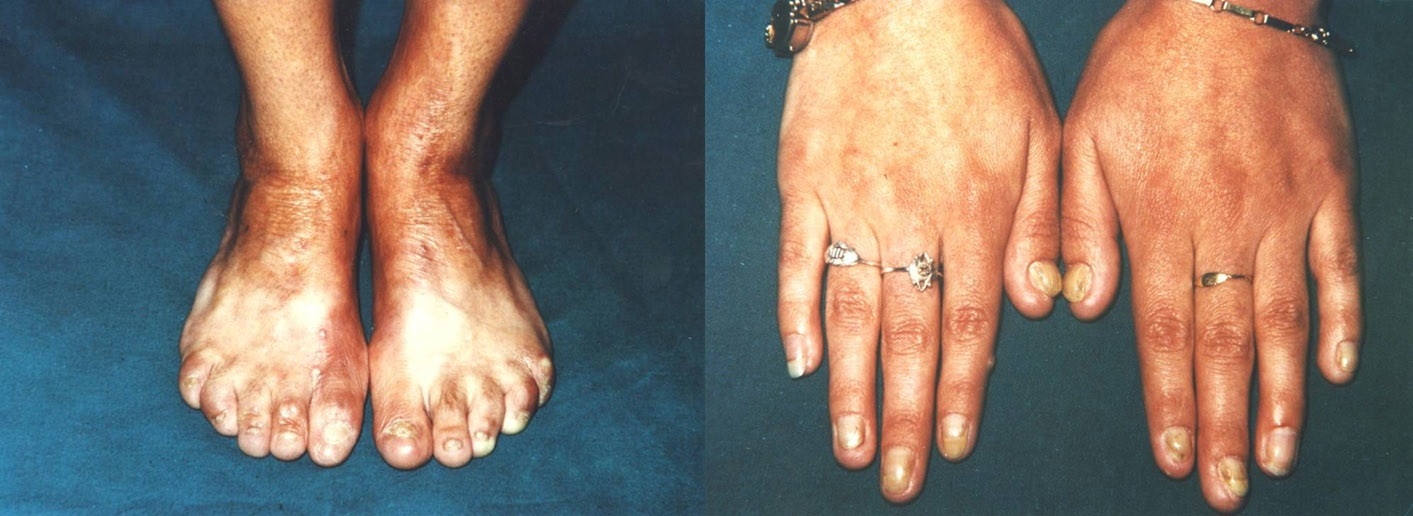
Case 2.—A 24-year-old woman with a history of blisters and erosions that developed at the sites of minor trauma from birth on was referred to our Institute. The patient was born with large, bilateral non-inflammatory, well demarcated, skin defects covered by red, thin, moist glistening epithelial membrane localized on the pretibial and inner lower legs, feet, including the plantar surface and dorsal aspects, and toes. The defect on the left leg extended above the knee. Slight bilateral dorsal flexion of the feet was seen at birth (Fig. 4). The denuded areas healed completely by growth of the skin from the borders, leaving a fine wrinkled scar within a few weeks.
 |  |
| Figure 4 | Figure 5 |
|---|---|
| Figure 4. Congenital localized absence of the skin in the patient with RDEB-I: bilateral sharply demarcated, clear-cut skin
defects localized on the lower legs, dorsal and plantar aspects of the toes. Figure 5. Figure 5. Remarkable nail changes characterized by partially thickened and curved fingernails and absent toenails in the patient with RDEB-I | |
Extensive blistering during the neonatal period was observed. Blisters and erosions at the trauma-prone skin or elsewhere after minor trauma or spontaneously were seen. During adolescence the disease activity diminished and the predilection for intertriginous sites developed. Blistering of the skin tended to be localized at the axillary area, perineum, lower part of the abdomen and lumbar area. Post-bullous atrophic hypo- and hyper-pigmented areas were noticed. Remarkable nail changes characterized by thickened and curved fingernails and absent toenails were present (Fig. 5). The teeth were highly carious. Blistering of the oral mucosa accompanied with difficulties in swallowing has been present since early childhood. Pronounced shortening of the lingual frenulum produced difficulty in protruding the tongue outside of the mouth. Dysphagia developed during adolescence. Esophageal stenosis was detected by radioscopy. The family history was negative for similar defects or evidence of blistering of the skin.
 |  |
| Figure 6a | Figure 6b |
|---|---|
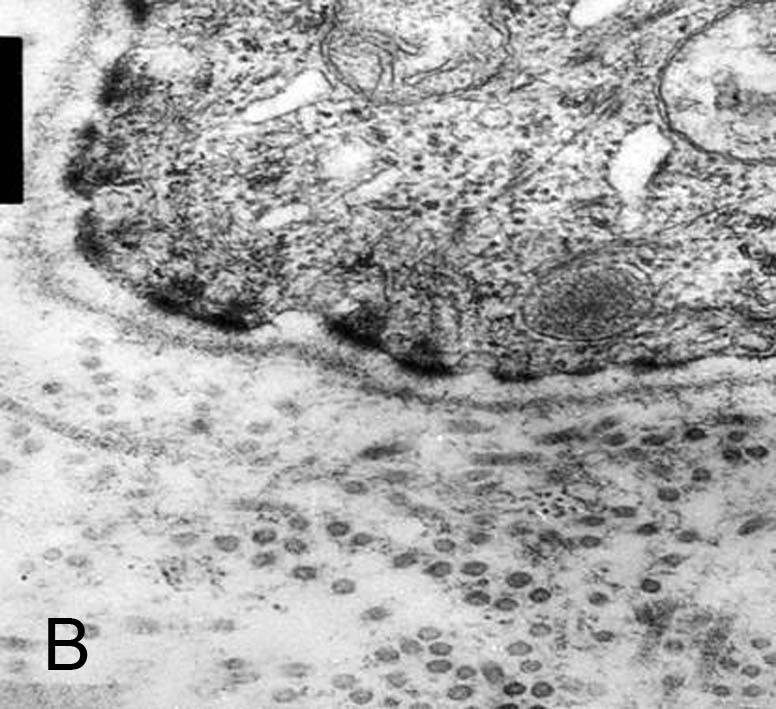
| Figure 6a. Electron micrograph showing dermolytic mode of blistering; blister formation takes place below the lamina densa.
In blister areas anchoring fibrils are absent (97,000X). Figure 6b. Rudimentary appearing anchoring fibrils are moderately to markedly decrease in the perilesional skin (119,000X). | |
Electron microscopy examination revealed cleavage beneath the level of lamina densa with the dermolytic mode of blistering (Fig. 6a). In the blister areas, along the epidermal roof of the blister anchoring fibrils were absent. In the blister floor pronounced collagenolysis was found. Anchoring fibrils were present, but markedly decreased and poorly formed along the epidermal roof of the blister and intact perilesional skin (Fig. 6b).
Discussion
Recessive dystrophic epidermolysis bullosa is a highly debilitating disorder displaying a spectrum of severe cutaneous and extracutaneous manifestations. The estimated incidence of RDEB in the United States in the period from 1986-1990 was 26 EB births per million live births [13]. Although different forms of EB have been linked to mutations in no less than 10 distinct genes encoding the major structural basement membrane zone proteins, both the autosomal dominant and recessive forms of dystrophic EB (DEB) result from mutations in the COL7A1 gene, encoding type VII collagen. This type of collagen is a major component of anchoring fibrils, structural elements that stabilize the attachment of the dermal-epidermal junction [14, 15].
Prenatal diagnosis of EB is necessary and requested in families at high risk for recurrence of EB. For more than two decades, fetoscopy has been successfully applied in the prenatal diagnosis of inherited EB. This technique obtains fetal skin biopsy specimens for ultrastructural analyses by transmission electron microscopy to identify possible structural defects in fetal skin [16, 17]. The fetoscopy is an invasive technique associated with a small risk of fetal mortality. The test must be performed relatively late, usually between 16 to 18 gestational weeks. Fetal skin biopsy has gradually been superseded by DNA-based diagnostic screening using fetal DNA from amniotic fluid cells (at 16-20 gestational week) or chorionic villi samples (at 10-12 gestational week) [18]. All cases of dystrophic EB result from mutations in a single gene, COL7A1. Preimplantation genetic diagnosis involving DNA analysis of single blastomeres extracted from late cleavage stage embryos following in vitro fertilization broadens the range of prenatal testing options [19]. Although molecular testing still remains primarily a research tool, it provides the additional information that can lead to the prenatal diagnosis in family affected with inherited EB.
Postnatal diagnosis is useful to confirm and establish the type of inherited EB and it involves the assessment of a combination of ultrastructural and antigenic features by transmission electron microscopy, immunofluorescence antigenic mapping, and EB-related monoclonal antibody studies. Skin biopsy of the affected child examined by electron microscopy allows identification of the level of skin cleavage and ultrastructural defects [20]. Immunofluorescence antigenic mapping involves modified indirect immunofluorescence using the patient's skin and antibodies against three known protein antigens located in dermoepidermal junction (bullous pemphigoid antigen in the upper portion of the lamina lucida, laminin in the lower portion of the lamina lucida and type IV collagen in the lamina densa) [21]. Monoclonal antibody studies are used to confirm the classification established by electron microscopy and immunofluorescence antigenic mapping. Several anti-basement membrane antibodies are used to subclassify types of EB (GB3, 19-DEJ-l, C6SPG, KF-1, LH 7:2, L3d, AF1/AF2).
The presentation of RDEB with ACC is very rare, with only five cases described in the published literature [5, 12]. In this paper we presented further two cases of RDEB presenting with ACC localized on the lower extremities.
In our patients with a negative family history and non-informative family pedigrees, molecular testing for meticulous postnatal diagnosis was not performed. Postnatal diagnosis in our study was confirmed by transmission electron microscopy findings and the presence of clinical features compatible with RDEB-HS and/or RDEB-nHS, following the algorithm proposed by Fine et al [2]. Electron microscopy examination in our study revealed a sublamina densa level of skin cleavage, which is consistent with the diagnosis of DEB for both our patients [2, 13]. Along with the sub-lamina densa split, which confirmed the dermolytic mode of blistering, complete absence of anchoring fibrils in patient No. 1 and reduced and rudimentary appearing anchoring fibrils in patient No. 2 were detected. Basic ultrastructural findings together with the specific clinical outcome characterized by marked growth retardation and severe cutaneous and extracutaneous disease involvement confirms the diagnosis of patient No. 1 as the major RDEB Hallopeau-Siemens subtype. The phenotype characterized by relatively localized blistering of the skin with a pronounced predilection for intertriginous sites in adulthood is consistent with a rare minor RDEB inversa subtype in patient No. 2.
Aplasia cutis congenita (ACC) is an uncommon group of disorders with an estimated incidence of approximately 1-3 in 10,000 births [3]. ACC may occur as an isolated defect or in association with other developmental anomalies and disorders. According to Freiden ACC presenting with simplex, junctional, or dystrophic EB is classified as ACC type 6 [4]. The diagnosis of ACC is primarily clinical, depending on the extension of compromise, mode of inheritance, and associated findings [3, 4]. The diagnosis of ACC was made clinically in our patients; biopsy of the skin defects was not performed. The clinical description of ACC in our patient confirmed ACC type 6 according to the Frieden classification [4].
ACC type 6 was initially described as Bart syndrome [5]. Bart syndrome, originally reported in a large family in 1966, was described as a new clinical entity consisting of congenital localized absence of skin (CLAS) on the lower extremities, non-scarring blistering of the skin and oral mucosa, and additional nail abnormalities [7]. Butler et al. were the first to report microscopic, ultrastructural and immunofluorescent mapping features on affected individuals with the complete inherited syndrome initially described by Bart [22]. They were also the first to document the association between dominant dystrophic EB and CLAS. Later on, Zelickson and coworkers reported the findings of clinical, ultrastructural, immuno-histologic, and genetic linkage studies of the original Bart's kindred and their descendants [23]. Analysis of Bart's original kindred demonstrated ultrastructural abnormalities in the anchoring fibrils and linkage of the inheritance of the disease to the region of chromosome 3 near the type VII collagen gene (COL7A1) [24]. Based on these findings Bart syndrome should be considered as a clinical variant of dominantly inherited dystrophic EB. In the last decade it is strongly recommended that the term, Bart syndrome, should be eliminated from future EB literature because CLAS may be an associated finding in any of the three major types of EB (simplex, junctional or dystrophic) [2].
Conclusions
A review of the literature clearly indicates that whenever ACC is associated with widespread skin blistering, careful clinical examination and continuous observation, along with a skin biopsy examined by transmission electron microscopy or immunofluorescence antigen mapping are necessary to reliably establish the diagnosis. Basic ultrastructural findings provided by transmission electron microscopic studies presented by sub-lamina densa split and complete absence or presence of rudimentary appearing anchoring fibrils along with clinical findings are sufficient for proper classification in DEB and further sub-classification in major RDEB-HS and RDEB-nHS, or minor RDB-I subtypes. Considerable overlap may occur in infancy and early childhood between RDEB subtypes. However, later on, predilection of the disease activity for intertriginous areas allowed sub-classification of an unclassified patient into RDEB inversa subtype. The diagnosis of ACC remains primarily clinical, depending on the extent of the involvement and associated findings.
References
1. Gedde-Dahl T Jr. Epidermolysis Bullosa. A clinical, genetic and epidemiologic study. Baltimore: The John Hopkins press, 1971; 1-180.2. Fine JD, Eady RA, Bauer EA, Briggaman RA, Bruckner-Tuderman L, Christiano A, Heagerty A, Hintner H, Jonkman MF, McGrath J, McGuire J, Moshell A, Shimizu H, Tadini G, Uitto J. .Revised classification system for inherited epidermolysis bullosa: report of the second International Consensus Meeting on diagnosing and classification of epidermolysis bullosa. J Am Acad Dermatol 2000; 42(6):1051-66. PubMed
3. Antaya R, Schaffer JV. Developmental Anomalies. In: Bologna JL, Jorizzo JL, Rapini RP (eds). Dermatology. New York: Mosby; 2003:915-931.
4. Frieden IJ. Aplasia cutis congenita: a clinical review and proposal for classification. J Am Acad Dermatol 1986; 14(4):646-60. PubMed
5. McCarthy MA, Clarke T, Powell FC. Epidermolysis bullosa and aplasia cutis. Int J Derm 1991; 30:481-484. PubMed
6. Bart BJ, Gorlin RJ, Anderson VE, Lynch FW. Congenital localized absence of skin and associated abnormalities resembling epidermolysis bullosa: a new syndrome. Arch. Derm 1966; 93:296-304. PubMed
7. Bart BJ. Epidermolysis bullosa and congenital localized absence of skin. Arch Derm 1970; 101:78-81. PubMed
8. Kanzler MH, Smoller B, Woodley DT. Congenital localized absence of the skin as a manifestation of epidermolysis bullosa. Arch Dermatol 1992; 128(8):1087-90. PubMed
9. Morrell DS, Rubenstein DS, Briggaman RA, et al. Congenital pyloric atresia in a newborn with extensive aplasia cutis congenita and epidermolysis bullosa simplex. Br J Dermatol 2000;143(6):1342-3. PubMed
10. Skoven, I, Drzewiecki KT. Congenital localized skin defect and epidermolysis bullosa hereditaria letalis. Acta Derm Venerol 1979; 59:533-537. PubMed
11. Al-Salem A, Nawaz A, Matta H, Jakobsz A. Congenital pyloric atresia: the spectrum. Int Surg 2002; 87(3):147-51. PubMed
12. Wojnarowska FT, Eady RA, Wells RS. Dystrophic epidermolysis bullosa presenting with congenital localized absence of skin: report of four cases. Br J Dermatol 1983; 108(4):477-83. PubMed
13. Fine JD, ed, Bauer EA, ed, McGuire J, ed, Moshell A, ed. Epidermolysis Bullosa: Clinical, Epidemiologic, and Laboratory Advances and the Findings of the National Epidermolysis Bullosa Registry. Baltimore, MD: Johns Hopkins University Press; 2000.
14. Uitto J, Pulkkinen L. Molecular genetics of heritable blistering disorders. Arch Dermatol 2001; 137(11):1458-61. PubMed
15. Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet 2007; 44(3):181-92. PubMed
16. Rodeck CH, Eady RA, Gosden CM. Prenatal diagnosis of epidermolysis bullosa lethalis. Lancet 1980; 1:949-952. PubMed
17. Holbrook KA, Fine JD, Elias S, Christiano AM. Prenatal diagnosis of inherited epidermolysis bullosa: ultrastructural, antigenic, and molecular approaches. In: Fine J-D, Bauer EA, McGuire J, Moshell A, editors. Epidermolysis bullosa: clinical, epidemiologic, and laboratory advances, and the findings of the National Epidermolysis Bullosa Registry. Baltimore: Johns Hopkins University Press; 1999. p. 351-73.
18. Fassihi H, Eady RA, Mellerio JE, Ashton GH, Dopping-Hepenstal PJ, Denyer JE, et al. Prenatal diagnosis for severe inherited skin disorders: 25 years' experience. Br J Dermatol 2006;154(1):106-13. PubMed
19. Fassihi H, Renwick PJ, Black C, McGrath JA. Single cell PCR amplification of microsatellites flanking the COL7A1 gene and suitability for preimplantation genetic diagnosis of Hallopeau-Siemens recessive dystrophic epidermolysis bullosa. J Dermatol Sci 2006;42(3):241-8. PubMed
20. Pfendner EG, Bruckner A, Conget P, Mellerio J, Palisson F, Lucky AW, Palisson F, Anne W. Lucky AW. Basic science of epidermolysis bullosa and diagnostic and molecular characterization: Proceedings of the 2nd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. International Journal of Dermatology 2007; 46:781-794. PubMed
21. Fine JD, Smith LT. Non-molecular diagnostic testing of inherited epidermolysis bullosa: current techniques, major findings, and relative sensitivity and specificity. In: Fine JD, Bauer EA, McGuire J and Moshell A, Editors, Epidermolysis bullosa: clinical, epidemiologic, and laboratory advances, and the findings of the National Epidermolysis Bullosa Registry, Johns Hopkins University Press, Baltimore (1999), pp. 48-78.
22. Butler DF, Berger TG, James WD, Smith TL, Stanely JR, Rodman OG. Bart's syndrome: microscopic, ultrastructural, and immunofluorescent mapping features. Pediatr Dermatol 1986; 3(2):113-8. PubMed
23. Zelickson B, Matsumura K, Kist D, Epstein EH Jr, Bart BJ. Bart's syndrome: ultrastructure and genetic linkage. Arch Derm 1995; 131: 663-668. PubMed
24. Christiano AM, Bart BJ, Epstein EH Jr, Uitto J. Genetic basis of Bart's syndrome: a glycine substitution mutation in the type VII collagen gene. J Invest Dermatol 1996; 106(6):1340-2. PubMed
© 2008 Dermatology Online Journal