Hyperimmunoglobulin E syndrome with a novel STAT3 mutation
Published Web Location
https://doi.org/10.5070/D31mt365wnMain Content
Hyperimmunoglobulin E syndrome with a novel STAT3 mutation
Robert Anolik MD, Sarina Elmariah PhD, Stephanie Lehrhoff MD, Henry J Votava MD, Frank T Martiniuk PhD, William Levis MD
Dermatology Online Journal 15 (8): 16
Department of Dermatology, New York UniversityAbstract
A 35-year-old man with severe eczematous dermatitis and recurrent staphylococcal skin infections, some of which required hospitalization, is presented. Other medical concerns include recurrent oral staphylococcal infections, otitis media, ocular herpes simplex virus keratitis, asthma, steroid-induced gastritis, steroid-induced cataracts, recurrent upper respiratory infections, and acute pharyngitis. Past medical history includes retained dentition of six primary teeth, two episodes of childhood pneumonia that required hospitalization, and three wrist and ankle fractures. Laboratory data showed an eosinophil count of 2,400 cells/ml; the highest IgE level was 17,028 IU/mL. Considering the clinical and laboratory findings, the diagnosis of hyperimmunoglobulin E syndrome was made. DNA sequencing showed a novel signal transducer and activator of transcription 3 (STAT3) gene mutation within intron 12, specifically adenine to cytosine, two base pairs upstream of exon 13.
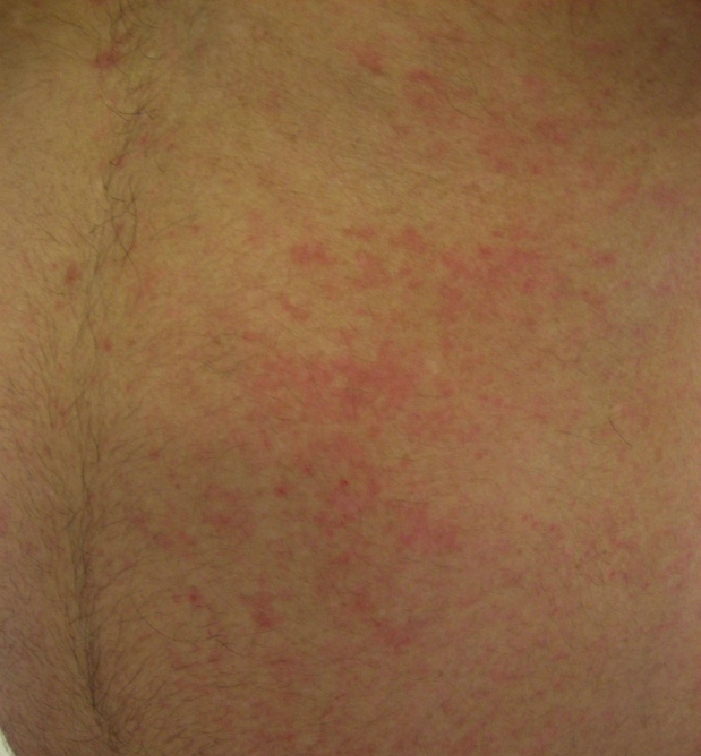 | 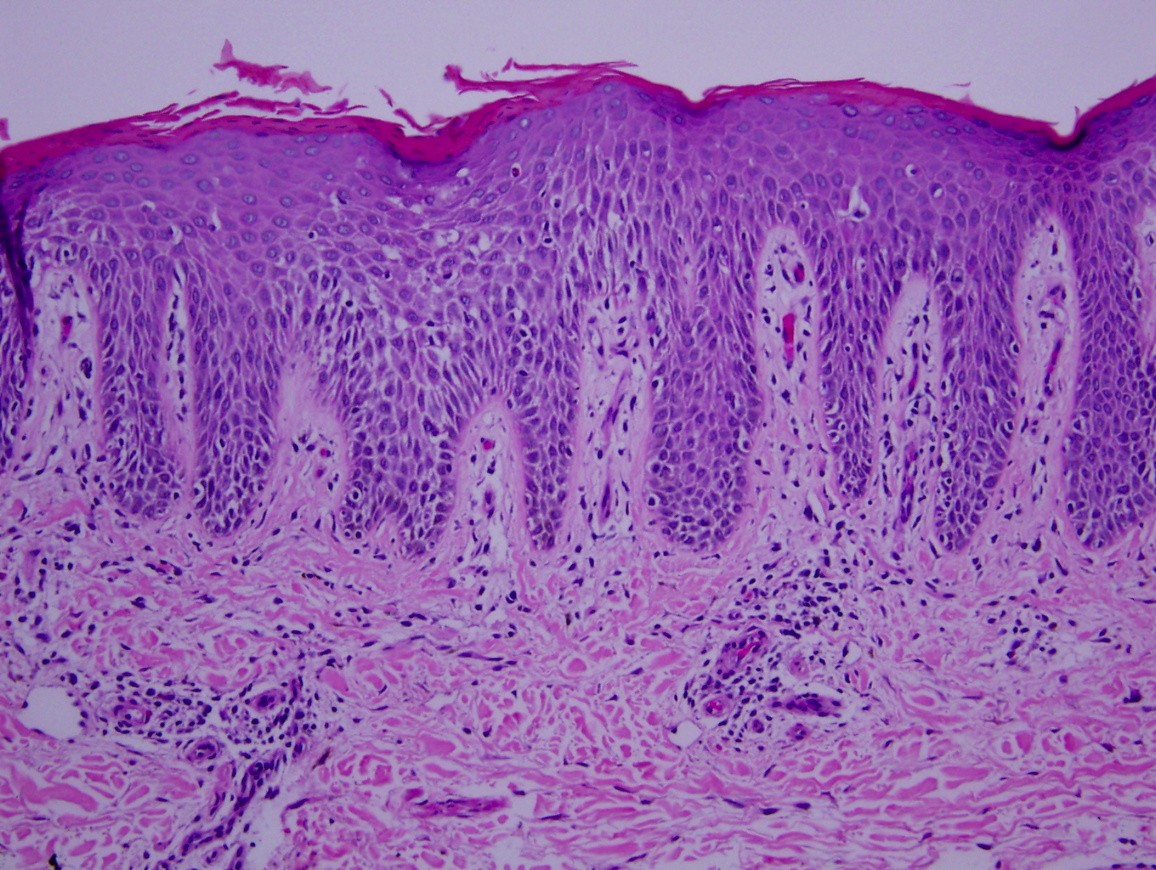 |
| Figure 1 | Figure 2 |
|---|---|
History
A 35-year-old man is managed in the Dermatology Clinic at Bellevue Hospital Center for a severe eczematous dermatitis and recurrent staphylococcal skin infections that at times have required hospitalization. Other hospital services, which include infectious disease, ophthalmology, and medicine, also are involved in his care.
Other medical concerns include recurrent oral staphylococcal infections, otitis media, herpes simplex virus keratitis, asthma, steroid-induced gastritis, steroid-induced cataracts, recurrent upper respiratory tract infections, and acute pharyngitis. Past medical history includes retained dentition of six primary teeth, two episodes of childhood pneumonia that required hospitalization, three wrist and ankle fractures, and an appendectomy. The patient recalls that he demonstrated joint hyperextensibility in his youth although this finding is not present today.
Medications include prednisone, azathioprine, hydroxyzine, tacrolimus 0.1 percent ointment, triamcinolone 0.1 percent ointment, valacyclovir, trimethoprim-sulfamethoxazole, albuterol, esomeprazole, risedronate, calcium supplements, and chlorhexidine gluconate 4 percent solution.
Physical Examination
Widespread, erythematous patches and thin, erythematosus, lichenified plaques with dry scale were present.
Laboratory data
The hemoglobin and hematocrit were normal. The eosinophil count was elevated at 5.7 percent. A basic metabolic panel was normal. Eosinophil counts are regularly elevated, with the highest count at 2,400 cells/ml. Immunoglobulin (Ig) E levels were chronically elevated, with the highest level at 17,028 IU/mL. IgA, IgM, and IgG levels were normal. Previous blood and skin cultures have been positive for methicillin-sensitive Staphylococcus aureus. Nares cultures have been negative. A human immunodeficiency virus screen was negative. A recent chest radiograph was normal. Scoliosis radiographic series demonstrated minimal dextroconvex thoracic scoliosis (less than 10 degrees) and minimal levoconvex lumbar curvature. A computed tomography (CT) scan of the face showed left periorbital fluid collection, with rim enhancement.
Histopathology
There is epidermal hyperplasia with spongiosis and mounds of parakeratosis. There is a superficial, perivascular infiltrate comprised of lymphocytes and scattered eosinophils.
Comment
In 1966, Davis, et al. first described the clinical triad of recurrent cold staphylococcal abscesses, sinopulmonary infections, and severe eczema as Job syndrome [1]. The syndrome is named for the biblical character Job: "so went Satan forth from the presence of Lord and smote Job with sore boils from the sole of his foot unto his crown." (Job, II, 7) Buckley, et al. later renamed the syndrome hyperimmunoglobulin E syndrome (HIES) after observing an elevated level of IgE in these patients [2].
Today, classic HIES is recognized as an autosomal-dominant or sporadic multisystem disorder that is characterized by high levels of IgE, sinopulmonary infections, and an eczematous dermatitis [3]. Other skin, bone, soft tissue, and immunologic abnormalities also are recognized that have variable and incomplete penetrance [3]. The National Institutes of Health (NIH) scoring system has been developed to assess HIES in patients and family members [4]. This system assesses common clinical findings in HIES: high levels of serum-IgE, eczematous dermatitis, recurrent skin abscesses, recurrent episodes of pneumonia, parenchymal lung anomalies, retained primary teeth, scoliosis, fractures with minor trauma, high eosinophil counts, course facies, midline anomalies, newborn eruptions, upper respiratory tract infections, candidiasis, other serious infections, fatal infection, hyperextensibility, lymphoma, increased nasal width, and high palate [4]. The scoring system includes a correction for age since many of these clinical findings are not present in early childhood [4]. Individual NIH scores can be variable, but some research models assign a threshold score of 40 for inclusion in study [5].
By applying the NIH scoring system to this patient, the diagnosis of HIES is more evident. His medical history launched his NIH score at 46, which is in the category of being affected [4]. His score is determined by assigning a score of zero for parenchymal lung anomalies (no high-resolution thoracic imaging was available), for scoliosis (his degree of scoliosis was too mild to qualify), for newborn eruption (the patient is unsure), and for absence of fatal infection, lymphoma, increased nasal width, high palate, with correction for young age. Importantly, the patient was assigned points for infectious processes that he experienced before starting immune modifying therapies.
Although the characteristic features of HIES have been described for decades, the etiology remained elusive. Recent developments, however, identified the causative genetic mutation. Cytokines are known to transduce signals through receptors and receptor-associated Janus kinases (JAKs) [6, 7]. JAKs phosphorylate signal transducer and activator of transcription (STAT) proteins, which then dimerize and translocate to the nucleus to activate target genes [6, 7]. While studying the JAK/STAT pathway in a single patient with an eczematous dermatitis, elevated IgE, and a mild T-cell deficiency, a homozygous mutation of tyrosine kinase 2, which is a member of the JAK family and activator of STAT4 protein, was identified [8]. Since this patient shared features with classic HIES, the JAK/STAT signaling pathway was more closely studied. Ultimately, in 2007, Minegishi, et al. and Holland, et al. independently identified the causative mutations in HIES, which are heterozygous mutations in the gene encoding STAT3 [9, 10].
Eight of fifteen unrelated HIES patients had heterozygous STAT3 mutations not found in their siblings or parents [9]. The peripheral blood of these patients showed altered responses to cytokines and diminished DNA binding ability of STAT3 [9]. Mutant STAT3 interaction with wild-type STAT3 in cell lines suggested a dominant- negative mechanism [9]. Cytokine phenotyping and DNA sequencing of STAT3 in 50 patients with HIES and 48 family members disclosed STAT3 mutations in either the DNA binding domain or Src homology 2 (SH2) in all patients affected with HIES [10]. Unlike the sporadic mutations in the Minegishi, et al. cohort, Holland, et al. observed in all patients affected with HIES both sporadic and autosomal-dominant patterns in multiple families studied [9, 10].
The role of STAT3 was reinforced when 37 of 38 additional HIES patients were described with STAT3 mutations [5]. Fourteen different STAT3 mutations were found, with most involving the previously described DNA binding domain and SH2 domain; however, new mutations were identified, which included intronic splice site mutations upstream from exon 12 [5].
Owing to the clinical diagnosis of HIES in the patient described in this report, a sequencing analysis of his STAT3 gene showed a novel mutation within intron 12, specifically adenine to cytosine, two base pairs upstream of exon 13. The previously reported STAT3 intronic mutations resulted in aberrant DNA splicing and subsequent in-frame deletions of amino acids [5]. It is possible this novel mutation of intron 12 yields similar abnormalities.
The multiorgan derangements of HIES are consistent with a mutation in STAT3. STAT3 has far-reaching effects, which include a key role in lung inflammation, osteoclast generation, peripheral eosinophilia, epithelial expression of beta-defensins, and certain cytokine signals, such as IL-6 and IL-10 [9, 11, 12, 13, 14]. Moreover, Renner, et al. highlight the dependence of Th17 cells on STAT3 function [5, 15, 16]. Th17 cells are integral in the recruitment, activation, and migration of neutrophils and therefore are critical in the clearance of fungal and extracellular bacterial infections [17, 18, 19]. Reduced or absent Th17 cells in the peripheral blood of HIES patients have been demonstrated [5]. Therefore, deficiencies of Th17 cells and IL-17 production likely contribute to the susceptibility to multiorgan infection.
The patient in this report represents a patient with HIES and a novel STAT3 mutation. Future studies for the patient include assessment of mRNA and cDNA. These studies are intended to clarify effects of the patient's intronic STAT3 mutation. Other steps will include enumeration of circulating Th17 cells.
References
1. Davis SD, et al. Job's syndrome: recurrent, "cold," staphylococcal abscesses. Lancet 1966; 1: 1013 [PubMed]2. Buckley RH, et al. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics 1972; 49: 59 [PubMed]
3. Grimbacher B, et al. Hyper-IgE syndrome with recurrent infection- an autosomal dominant multisystem disorder. N Engl J Med 1999; 340: 692 [PubMed]
4. Grimbacher B, et al. Genetic linkage of hyper-IgE syndrome to chromosome 4. Am J Hum Genet 1999; 65: 735 [PubMed]
5. Renner ED, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced Th17 cell numbers, and variable defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol 2008; 122: 181 [PubMed]
6. Schindler C, et al. Transcriptional responses to polypeptide ligands; the JAK- STAT pathway. Annu Rev Biochem 1995; 64: 621 [PubMed]
7. Ihle, JN. Cytokine receptor signaling. Nature 1995; 377: 591 [PubMed]
8. Minegishi Y, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 2006; 25: 745 [PubMed]
9. Minegishi Y, et al. Dominant negative mutation in DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007; 448: 1058 [PubMed]
10. Holland S, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 2007; 357: 1608 [PubMed]
11. Hokuto I, et al. Stat-3 is required for pulmonary homeostasis during hyperoxia. J Clin Invest 2004; 113: 28 [PubMed]
12. Welte T, et al. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of TAT3 in innate immunity. Proc Natl Acad Sci USA 2003; 100: 1879 [PubMed]
13. Zhange Z, et al. Osteoporosis with increased osteoclastogenesis in hematopoietic cell-specific STAT3-deficient mice. Biochem Biophys Res Commun 2005; 328: 800 [PubMed]
14. Wolk K, et al. IL-22 increases the innate immunity of tissues. Immunity 2004; 21: 241 [PubMed]
15. Milner JD, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008; 452: 773 [PubMed]
16. Mathur AN, et al. Stat3 and Stat4 direct development of IL-17 secreting Th cells. J Immunol 2007; 178: 4901 [PubMed]
17. Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med 2007; 13: 139 [PubMed]
18. Huang W, et al. Requirement of interleukin-17A for systemic anti-candida albicans host defense in mice. J Infect Dis 2004; 190: 624 [PubMed]
19. Acosta-Rodriguez, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 2007; 8: 639 [PubMed]
© 2009 Dermatology Online Journal