Two pediatric cases of Blau syndrome
Published Web Location
https://doi.org/10.5070/D31gz33233Main Content
Two pediatric cases of Blau syndrome
Donald A Glass II MD, PhD1, Jennifer Maender MD1, Denise Metry MD1,2
Dermatology Online Journal 15 (12): 5
1. Department of Dermatology, Texas Children's Hospital, Baylor College of Medicine, Houston, Texas2. Department of Pediatrics, Texas Children's Hospital, Baylor College of Medicine, Houston, Texas
Case reports
Patient 1. A 5-year-old African American male was referred to pediatric dermatology for the evaluation of a generalized non-pruritic skin eruption. This began at 2.5 years of age and was initially diagnosed as eczema, but failed to respond to topical steroid therapy. At age 3 years, he developed edema and mild pain of his wrists, knees, and ankles, at which time he was diagnosed with juvenile rheumatoid arthritis (JRA). After a failed kindergarten visual screening examination, his mother was notified that a formal ophthalmologic evaluation was needed. His medications at presentation included methotrexate, methylprednisolone, and naproxen. There was no family history of similar conditions.
 |  |
| Figure 1 | Figure 2 |
|---|---|
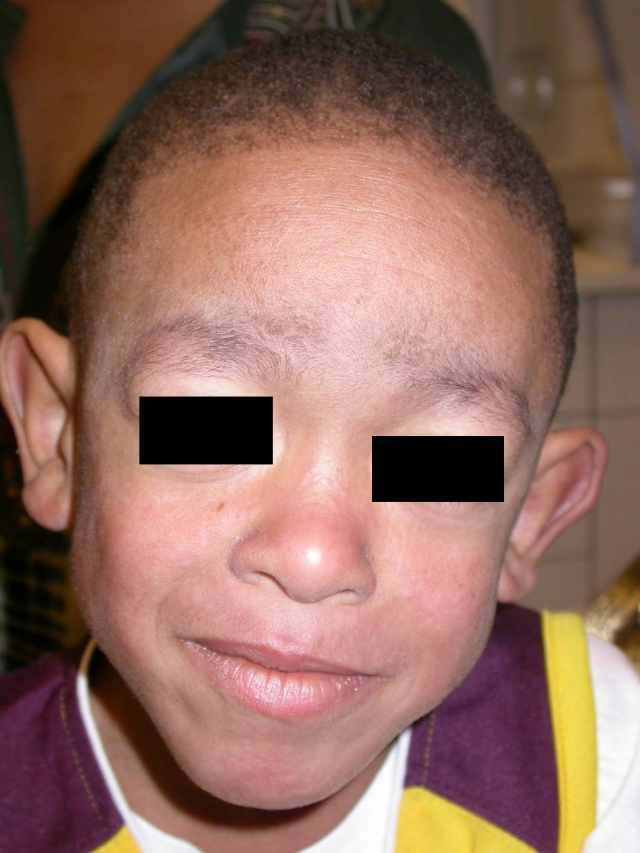
| Figure 1. Multiple, reddish-brown papules coalescing over the right arm in a boy with Blau syndrome Figure 2. Coarse facial features in a boy with Blau syndrome | |
Physical examination showed generalized, red to violaceous flat-topped papules, most prominent over his extensors (Fig. 1). His face had a coarse appearance without overt papules (Fig. 2). He had significant boggy edema of his wrists with lesser involvement of his knees. His sclerae appeared mildly injected.
 |  |
| Figure 3 | Figure 4 |
|---|---|
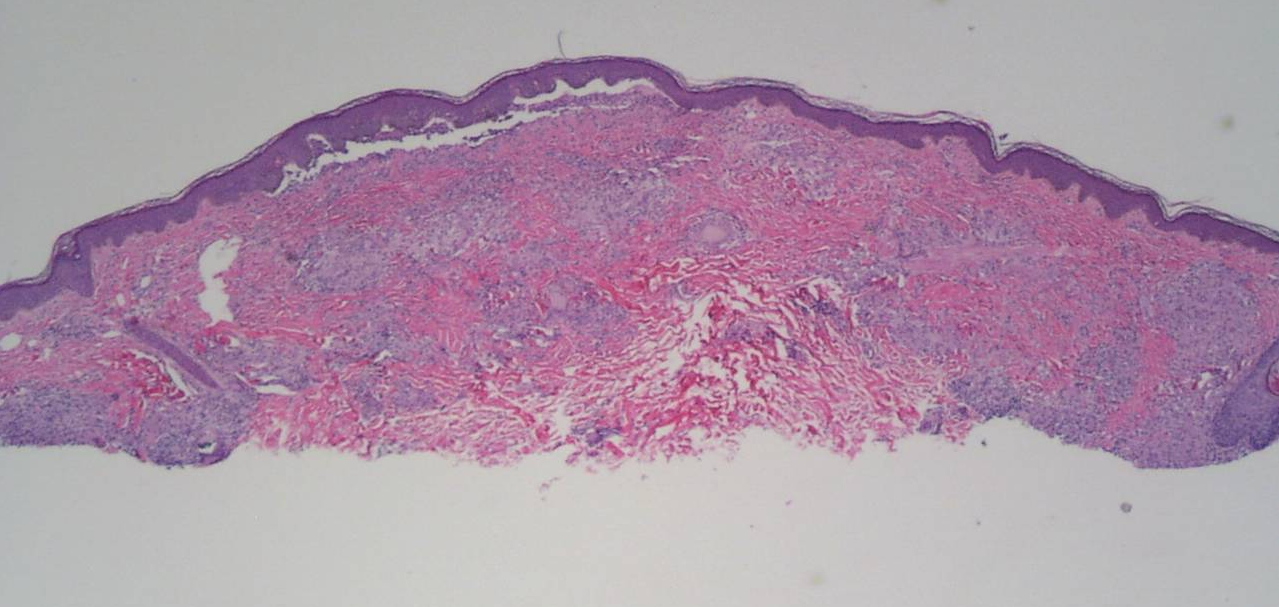
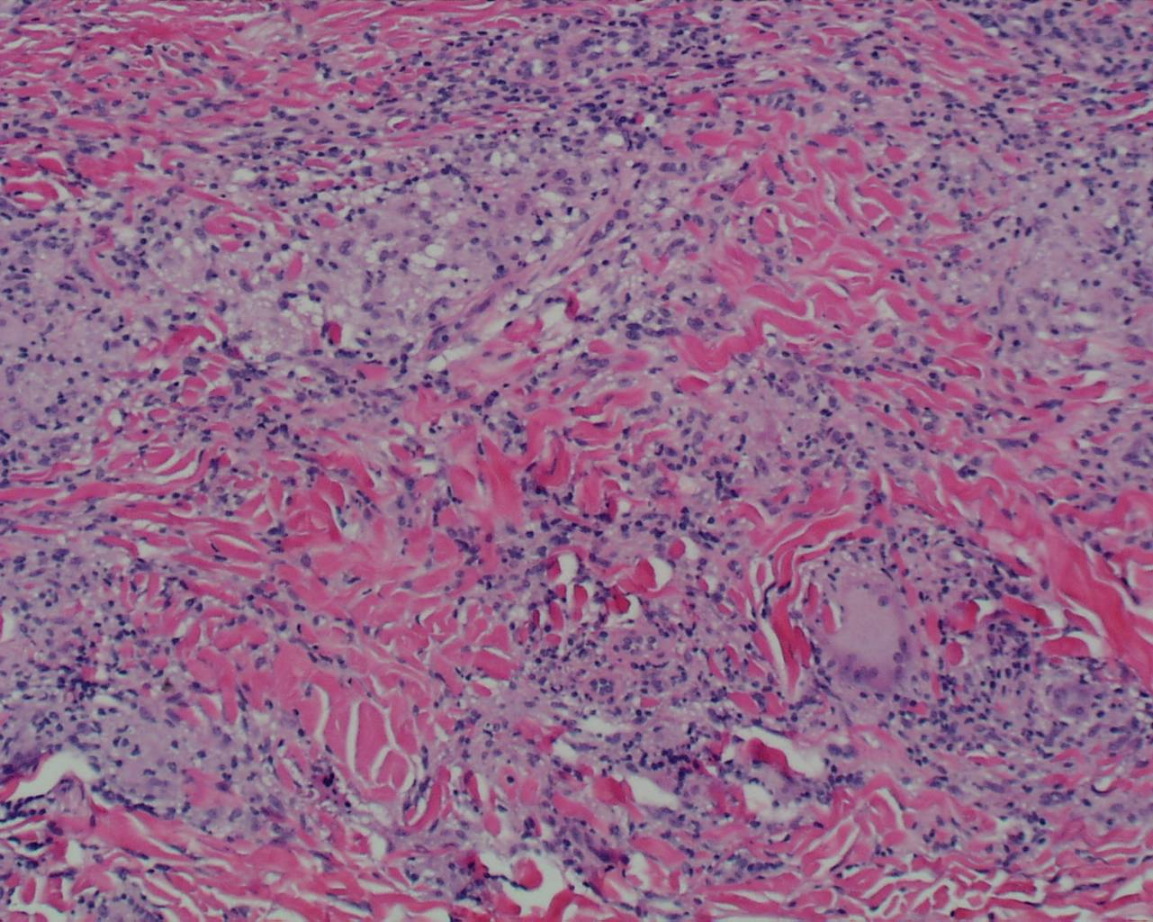
| Figure 3. Skin biopsy shows multiple non-caseating granulomas throughout the dermis. (H&E, x100) Figure 4. Non-caseating granulomas with enlarged epithelioid histiocytes and multinucleated giant cells. (H&E, x400) | |
A skin biopsy taken from a papule on his right arm showed multiple non-caseating granulomas throughout the dermis (Fig. 3), characterized by enlarged epithelioid histiocytes and multinucleated giant cells. (Fig. 4) Acid-fast bacterial (AFB) and fungal stains were negative. A rheumatoid factor (RF) was reactive. He was referred to pediatric ophthalmology, where he was diagnosed with anterior uveitis, posterior synechiae with pupillary membrane, and posterior subcapsular cataracts. He was also referred for genetics evaluation, but his mother chose not to pursue testing for the CARD-15 gene.
Patient 2. An 8-year-old Hispanic male was referred for the evaluation of skin lesions in association with chronic arthritis and uveitis. He had a history of joint pain and swelling that first developed at 6 months of age, at which time he was diagnosed with JRA in Mexico and treated with systemic corticosteroids, with mild improvement. By age 2 years, he developed a generalized papular eruption that failed to respond to topical corticosteroids. The patient and his family later immigrated to the United States, where he was re-evaluated for JRA and started on treatment with methotrexate, prednisone, and etanercept, with only minimal improvement.
The child then presented to pediatric dermatology with a 6-month history of a generalized papular skin eruption, and the recent added diagnosis of chronic uveitis. His medications at presentation included fluorometholone and atropine ophthalmic drops, naproxen, methotrexate, and etanercept. Family history was significant for severe deforming arthritis in the patient's mother, and a younger brother with chronic arthritis and uveitis but no skin lesions.
Physical examination showed generalized skin-colored papules, most prominent on his abdomen. He had ulnar deviation of his hands and external rotation of his feet.
Pertinent laboratory evaluations included an angiotensin converting enzyme level of 86 mU/mL (nl = 15-60), erythrocyte sedimentation rate of 23 mm/hr (nl = 0-20), and elevated IgG level of 1970 (nl = 635-1284 mg/dL). His RF was nonreactive. Serum calcium and 24-hr urine calcium excretion were normal. A chest X-ray was normal, and radiographs of his knees showed bilateral osteopenia, a likely result of long-term systemic corticosteroid use. A skin biopsy obtained from a papule on his abdomen showed noncaseating granulomatous inflammation. AFB and fungal stains were negative. The patient, his brother, and his mother all tested positively for CARD 15 gene mutations.
Discussion
In 1985 Blau et al. described the presence of graulomatous arthritis, iritis, and skin lesions in 11 family members spanning four generations [1]. Similar findings of granulomatous synovitis and uveitis were described by Jabs et al [2]. The term Blau syndrome (OMIM# 186580) was first proposed by Pastores et al [3].
Blau syndrome, also known as familial juvenile systemic granulomatosis or Jabs Syndrome, is characterized by asymptomatic skin lesions of varying morphology that have been described as eczematous, ichthyosiform, and lichenoid. Characteristic systemic findings include large, boggy synovial effusions and cysts, anterior uveitis and focal posterior synechiae. Flexion contractures of the fingers and toes (camptodactyly) have also been reported. Notably, affected patients do not have the typical findings of bilateral hilar adenopathy and/or interstitial fibrosis typical of adult-onset sarcoidosis [4]. Blau syndrome has variable expressivity and usually affects preschool age children younger than four years of age.
Several reports have described other unusual associations with Blau syndrome. Liver and renal interstitial granulomata have each been described in one member of a family [5, 6]. Another family with Blau syndrome was found to have ichthyosis vulgaris [7]. There have also been reports of central nervous system involvement, including sixth nerve palsies and corticosteroid-responsive hearing loss [2, 8]. Another patient with large recalcitrant leg ulcers, in which biopsies taken from the ulcers consistently showed granulomas, was recently described [9].
Common laboratory abnormalities associated with the syndrome include hypercalcemia, hypercalciuria, elevated angiotensin converting enzyme (ACE) level, elevated erythrocyte sedimentation rate (ESR), elevated immunoglobulins, leukopenia, eosinophilia, hematuria, proteinuria, pyuria, and abnormal liver function tests [4, 10] Rheumatoid factor is often positive, a finding not typical of patients with JRA.
Histopathology shows ill-defined, superficial, dermal granulomatous inflammation. The granulomas contain epithelioid cells, occasional Langerhans-type multinucleated giant cells, and a few lymphoid cells, without evidence of necrosis. Perivascular lymphomononuclear infiltration around dermal vasculature is also typical.
Blau syndrome is clinically indistinguishable from early-onset sarcoidosis, both of which feature arthritis, uveitis, and dermatitis. In addition, the genetic mutation for both has been shown to involve the CARD15/NOD2 gene on chromosome16q12 [11]. CARD15 plays a role in the activation of nuclear factor kappa B, an up-regulator of the transcription of pro-inflammatory cytokines. While the mutations in Blau syndrome are autosomal dominantly inherited, early-onset sarcoidosis arises from de novo mutations in CARD15 [12, 13, 14]. Both Blau syndrome and early-onset sarcoidosis seemingly arise from a different mechanism than adult-onset sarcoidosis, which is unassociated with CARD 15 mutations [15, 16]. While CARD15 mutations are associated with Crohn disease, which also features granulomatous arthritis and uveitis, such mutations occur at a different domain within CARD15 than Blau syndrome. Furthermore, other features of Crohn, notably gut inflammation and pyoderma gangrenosum, are absent in Blau syndrome and early-onset sarcoidosis [17, 18].
The initial presentation of joint manifestations in the absence of ocular or skin findings is often mistaken for JRA. However, Blau syndrome has milder constitutional symptoms and characteristic joint abnormalities not seen in JRA. The diagnosis can be suppported by the demonstration of noncaseating granulomas within skin, synovial or conjunctival biopsies, and the presence of CARD15/NOD2 mutations. Any patient with a diagnosis of JRA and a family history of granulomatous disease and/or multifocal choroiditis should be evaluated for Blau syndrome [19].
Systemic corticosteroids are the mainstay of therapy; however, given the chronicity of this disease and the young age of onset, steroid-sparing agents such as methotrexate are often used for control of arthritis, progressive lung disease, disfiguring skin lesions, myocardial involvement, renal insufficiency, eye disease, neurological disease and/or abnormal calcium metabolism [4]. Case reports of the use of TNF-α inhibitors have thus far yielded mixed results [20, 21, 22]. Uveitis can be successfully treated with hourly corticosteroid ophthalmic drops. Calcium metabolism should also be monitored by measuring serum calcium and 24-hr urinary calcium excretion.
The prognosis for patients with Blau syndrome is one of any chronic, progressive, debilitating disease. With proper treatment, the sequelae of chronic arthritis and uveitis can be lessened, but as with many chronic conditions, the side effects of long-term systemic therapies may prove as disabling as the condition itself.
References
1. Blau EB. Familial granulomatous arthritis, iritis and rash. J. Pediat. 1985;107:689-693. [PubMed]2. Jabs DA et al. Familial granulomatous synovitis, uveitis, and cranial neuropathies. Am. J. Med. 1985;78:801-804. [PubMed]
3. Pastores GM et al. Autosomal dominant granulomatous arthritis, uveitis, skin rash and synovial cysts. J. Pediat. 1990;117:403-408. [PubMed]
4. Pattishell EN et al. Childhood Sarcoidosis. Pediatrics 1986;108:169-177. [PubMed]
5. Saini SK and Rose CD. Liver involvement in familial granulomatous arthritis (Blau syndrome). J. Rheum.1996;23:396-399. [PubMed]
6. Ting SS et al. Familial granulomatous arthritis (Blau syndrome) with granulomatous renal lesions. J Pediatr. 1998;133:450-452. [PubMed]
7. Masel G and Halbert A. Blau syndrome presenting with ichthyosis. Australas J Dermatol. 2005;46:29-32. [PubMed]
8. Emaminia A, et al. Central nervous system involvement in Blau syndrome: a new feature of the syndrome? J Rheumatol. 2007;34:2504-2505. [PubMed]
9. Dhondt V, et al. Leg ulcers: a new symptom of Blau syndrome? Eur J Dermatol. 2008;35:943.
10. Rodriguez GE et al. Serum angiotensin-converting enzyme activity in normal children and in those with sarcoidosis. J Pediatr. 1981;99:68-72. [PubMed]
11. Miceli-Richard et al. CARD15 mutations in Blau syndrome. Nature Genet. 2001;29:19-20. [PubMed]
12. Kanazawa N et al. Presence of a sporadic case of systemic granulomatosis syndrome with a CARD15 mutation. J. Invest. Derm. 2004;122:851-852. [PubMed]
13. Rose CD et al. Blau syndrome mutation of CARD15/NOD2 in sporadic early onset granulomatous arthritis. J Rheumatol. 2005;32:373-375. [PubMed]
14. Priori R et al. Sporadic Blau syndrome with a double CARD15 mutation. Report of a case with lifelong follow-up. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21:228-231. [PubMed]
15. Schurmann M et al. CARD15 gene mutations in sarcoidosis. Eur Respir J. 2003;22:748-754. [PubMed]
16. Martin TM et al. Uveitis in patients with sarcoidosis is not associated with mutations in NOD2 (CARD15). Am J Ophthalmol. 2003;136:933-935. [PubMed]
17. Hugot JP et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599-603. [PubMed]
18. Ogura Y et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603-606. [PubMed]
19. Latkany PA et al. Multifocal choroiditis in patients with familial juvenile systemic granulomatosis. Am J Ophthalmol. 2002;134:897-904. [PubMed]
20. Cuesta IA et al. Blau Syndrome (Familial granulomatous arthritis, iritis, and rash) in an African-American family. J Clin Rheumatol. 2000; 6:30-34. [PubMed]
21. de Oliveira SK et al. Indications and adverse events with the use of anti-TNF alpha agents in pediatric rheumatology: experience of a single center. Acta Reumatol Port. 2007;32(2):139-50. [PubMed]
22. Milman N et al. Favourable effect of TNF-alpha inhibitor (infliximab) on Blau syndrome in monozygotic twins with a de novo CARD15 mutation. APMIS. 2006;114(12):912-9. [PubMed]
© 2009 Dermatology Online Journal