Multiple glomus tumors and segmental neurofibromatosis: There are no coincidences
Published Web Location
https://doi.org/10.5070/D31c22g647Main Content
Multiple glomus tumors and segmental neurofibromatosis: There are no coincidences
Rita Cabral, F Santiago, O Tellechea
Dermatology Online Journal 17 (3): 4
Hospitais da Universidade de CoimbraAbstract
Segmental neurofibromatosis is a rare subtype of neurofibromatosis type 1 (NF1). Glomus tumors are uncommon benign tumors. The authors report the association between these two rare conditions, not yet reported.
Introduction
Segmental neurofibromatosis (SN) is a rare subtype of neurofibromatosis type 1 (NF1). As is true in other mosaic phenomena, it has variable phenotypes, with cutaneous and neurologic manifestations restricted to a specific area of the body, with no family history. Segmental neurofibromatosis results from a postzygotic mutation that occasionally may involve the gonads [1]. Cutaneous neurofibromas (CN), café-au-lait macules (CALMs), or both, in one or more dermatomes, are the main lesions that can be found. CNs are the most frequent presentation consisting of fibroblasts, mast cells, perineural cells, axons, and Schwann cells, arising from the neural crest [2]. Café-au-lait macules can be distributed or grouped along Blaschko lines.
Glomus tumors are uncommon benign tumors that originate in the neuromyoarterial elements of the glomus body, an arteriovenous shunt specialized in thermoregulation. Multiple glomangiomas or glomuvenous malformations are less common than the solitary variant and exhibit particular clinical and histopathological characteristics. They are inherited in an autosomal dominant pattern and appear as small red-blue papules or nodules (typically <5 mm), which are multifocal, painless or slightly painful, and present at birth in many cases.
Case report
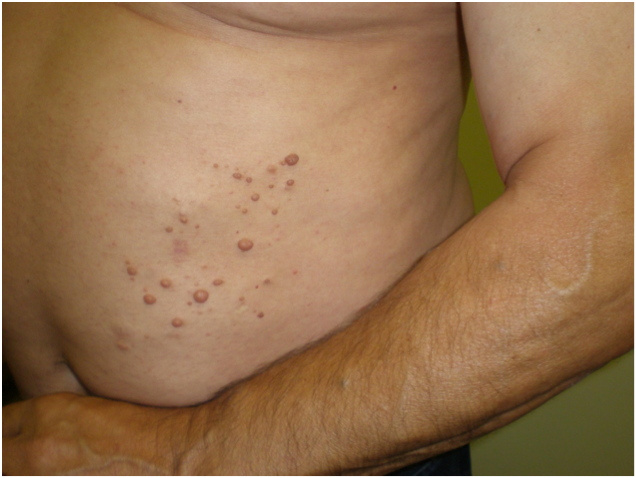 |
| Figure 1 |
|---|
We report the case of a 62-year-old male with multiple dome-shaped papules, skin colored and painless that were 3-5 mm in size. These were grouped in the left abdominal area and had evolved over 20 years. The patient also presented with multiple round blue papules that were soft, subcutaneous, and painless. These were distributed along the arms and trunk (Figure 1). The latter lesions had progressively appeared since adolescence. The patient had a family history of these last described lesions (mother, sister, and daughter), in an apparently hereditary autosomal dominant pattern.
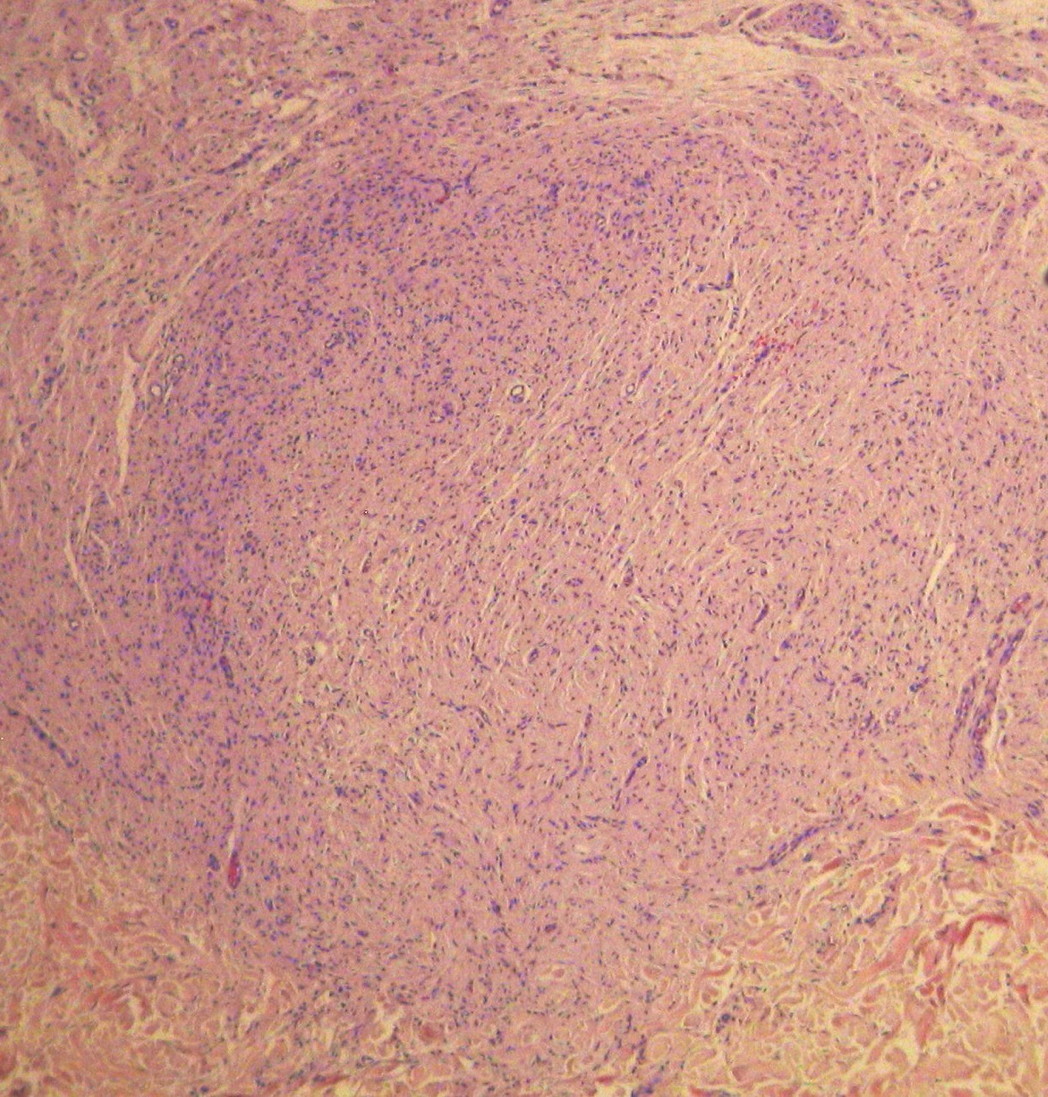 | 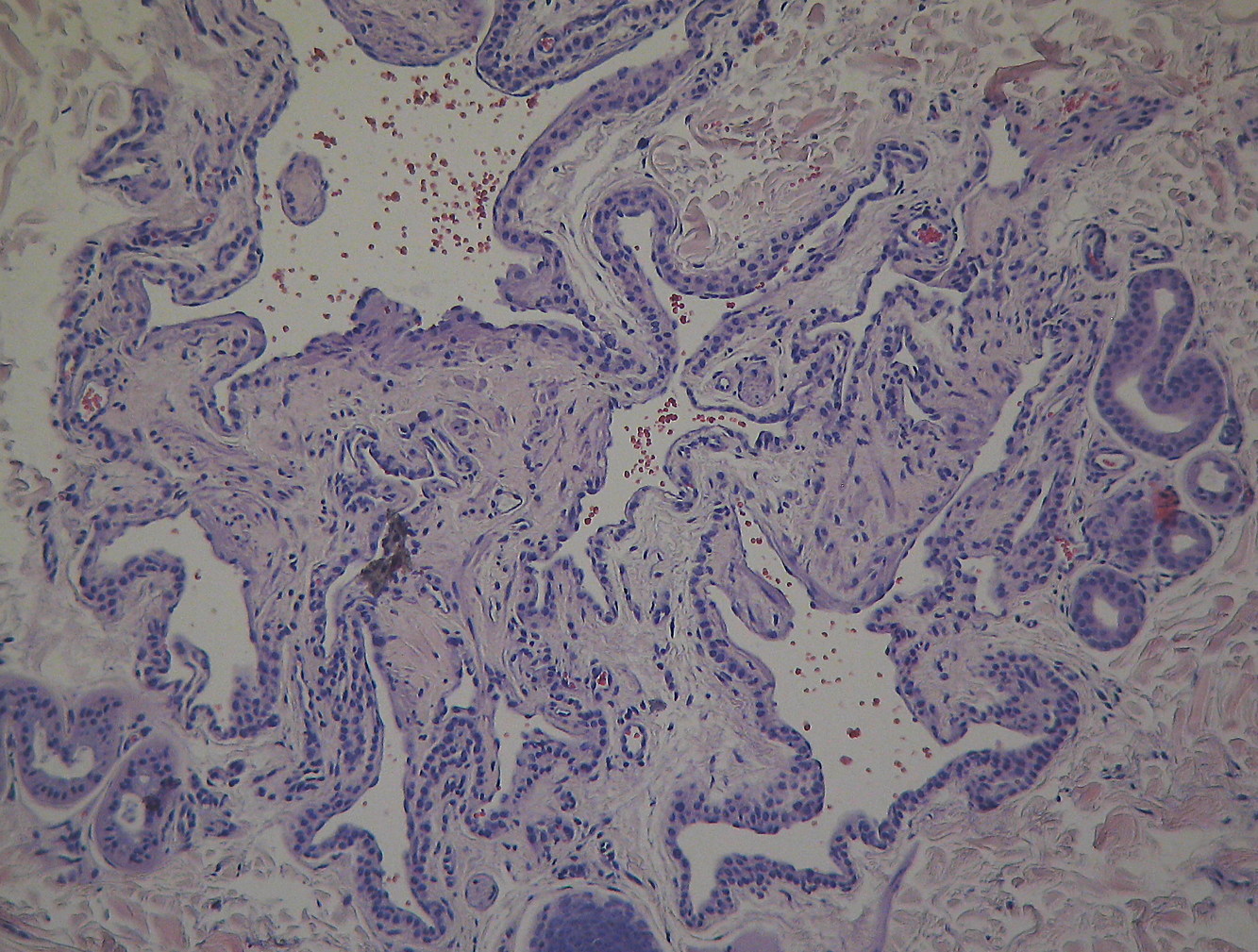 |
| Figure 2a | Figure 2b |
|---|
Histopathology examination of the first type of lesion identified a neurofibroma. The latter was identified as a glomangioma (Figures 2a and 2b).
Discussion
After review of the literature, we could verify that, although rarely reported, there is a well established relationship between NF1 and glomus tumors, single or multiple. The occurrence of glomus tumors, single or multiple, in patients with NF1 does not appear to be fortuitous.
De Smet et al [2] and Brems et al [5] postulated that the glomus cell (the tumor cell in glomus tumors) is of neural crest origin, just like Schwann cells (the tumor cells in neurofibromas). This hypothesis takes into account an origin in the pluripotent stem cells of the neural crest that can form three distinct cellular lines: neurons, Schwann cells, and smooth muscle-like myofibroblasts positive for αSMA, just like the αSMA positive glomus cells in the glomus tumors. Only αSMA-positive glomus cells harbor somatic and germline NF1 mutations. In addition, a similar two-hit mechanism, with bi-allelic inactivation of the tumor suppressor gene NF1 in αSMA-positive glomus cells, just like the one described in neurofibroma-derived Schwann cells, could explain an increased rate of these tumors in NF1 patients [5].
Our report stands out because it describes the association between two rare conditions - SN and glomangiomas - not yet reported. The only similar case was described by Brems et al [5], but it reported the association of a single, distal glomus tumor and SN.
References
1. Boyd KP, Korf BR, Theos A .Neurofibromatosis type 1. J Am Acad Dermatol 2009 Jul; 61(1): 1-14; quiz 15-6. [PubMed]2. De Smet L, Sciot R, Legius E. Multifocal glomus tumours of the fingers in two patients with neurofibromatosis type 1. J Med Genet 2002 Aug; 39(8):e45. [PubMed]
3. Rutkowski JL, Wu K, Gutmann DH, Boyer PJ, Legius E. Genetic and cellular defects contributing to benign tumor formation in neurofibromatosis type 1. Hum Mol Genet 2000 Apr 12; 9 (7):1059-66. [PubMed]
4. Klaber R. Morbus Recklinghausen with glomoid tumours. Proc R Soc Med 1938 Feb; 31(4): 347. [PubMed]
5. Brems H, Park C, Maertens O et al. Glomus tumors in Neurofibromatosis Type 1: Genetic, Functional, and Clinical Evidence of a novel association. Cancer Res 2009 Oct 15; 69 (20): 8216. [PubMed]
6. Sawada S, Honda M, Kamide R, Niimura M. Three cases of subungual glomus tumors with von Recklinghausen neurofibromatosis. J Am Acad Dermatol 1995 Feb; 32 (2 Pt1): 277-8. [PubMed]
7. Masuda T, Ishibashi Y, Tokuda Y. Multiple glomus tumors. Jpn J Dermatol 1962 Apr; 72:325-36. [PubMed]
8. Okada O, Demitsu T, Manabe M, Yoneda K. A case of multiple subungual glomus tumors associated with neurofibromatosis type 1. J Dermatol 1999 Aug; 26(8): 535-7. [PubMed]
© 2011 Dermatology Online Journal