Langerhans Cell Histiocytosis: A case report and summary of the current recommendations of the Histiocyte Society
Published Web Location
https://doi.org/10.5070/D317b778j6Main Content
Langerhans Cell Histiocytosis: A case report and summary of the current recommendations of the Histiocyte Society
Elizabeth K Satter MD1, Whitney A High MD2
Dermatology Online Journal 14 (3): 3
1. Departments of Dermatology and Dermatopathology, Naval Medical Center, San Diego, CA. Elizabeth.Satter@med.navy.mil 2.
Assistant Professor, Dermatology and Dermatopathology, University of Colorado Heath Sciences Center, Denver, ColoradoAbstract
Langerhans cell histiocytosis (LCH) is a rare proliferative disorder in which pathological Langerhans cells (LCs) accumulate in a variety of organs. Historically, the nomenclature regarding LCH has been confusing because the disease had been sub-categorized simply based upon different clinical manifestations. Herein, we report a child with the classic finding of disseminated LCH and summarize the current recommendation of the Histiocyte Society regarding the classification, evaluation, prognosis and treatment of LCH.
Introduction
Langerhans cell histiocytosis (LCH) represents a heterogeneous group of disorders, with presentations ranging from the fulminant, to those that spontaneously involute without any lasting sequelae. Historically, eponymous nomenclature was used to segregate amongst these varying presentations, but with the advent of ultrastructural studies and immunohistochemical staining techniques, most have been unified under the rubric of LCH. Herein we report a child with disseminated LCH who presented with failure to thrive, diabetes insipidus, osteolytic bony lesions and an extensive cutaneous eruption. Through this pedagogic case, we summarize the current recommendations of the Histiocyte Society regarding the classification, evaluation, prognosis, and treatment of LCH.
Case Report
A 3-year-old boy was admitted to the Medical University of South Carolina Children's Hospital for failure to thrive. The child was small for age, cachetic, and mildly lethargic. Progressive weight loss had been noticed over the last year, which was accompanied by vomiting, polyuria, and polydipsia. During this time, an extensive cutaneous eruption also became apparent.
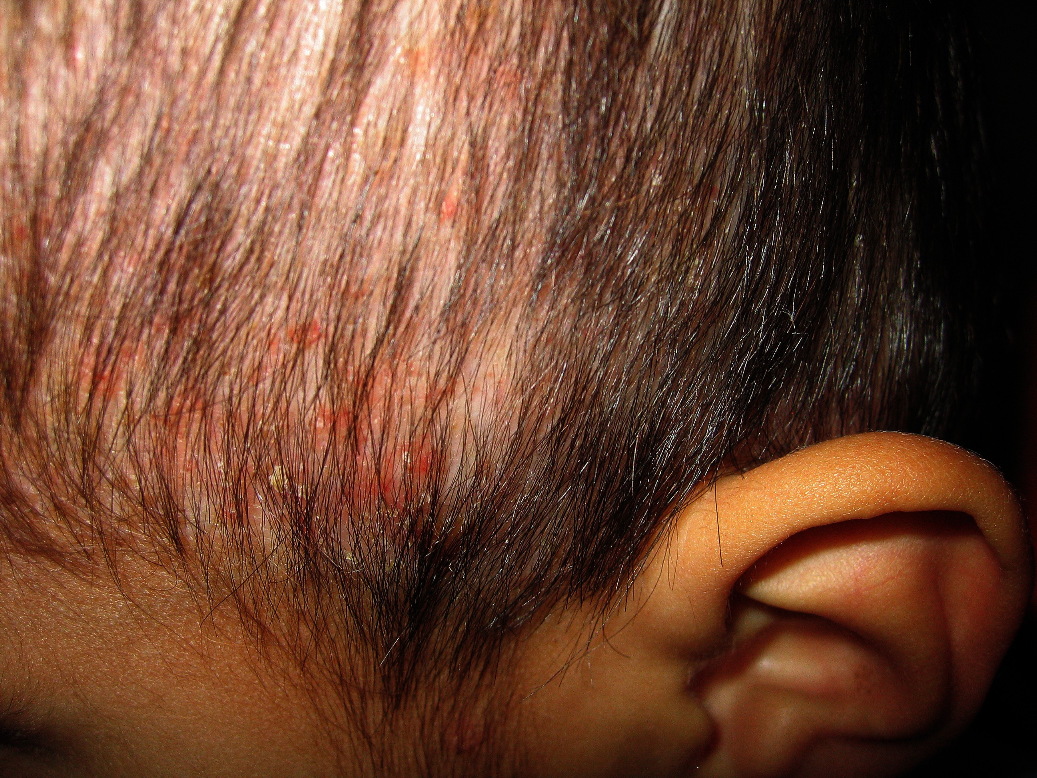
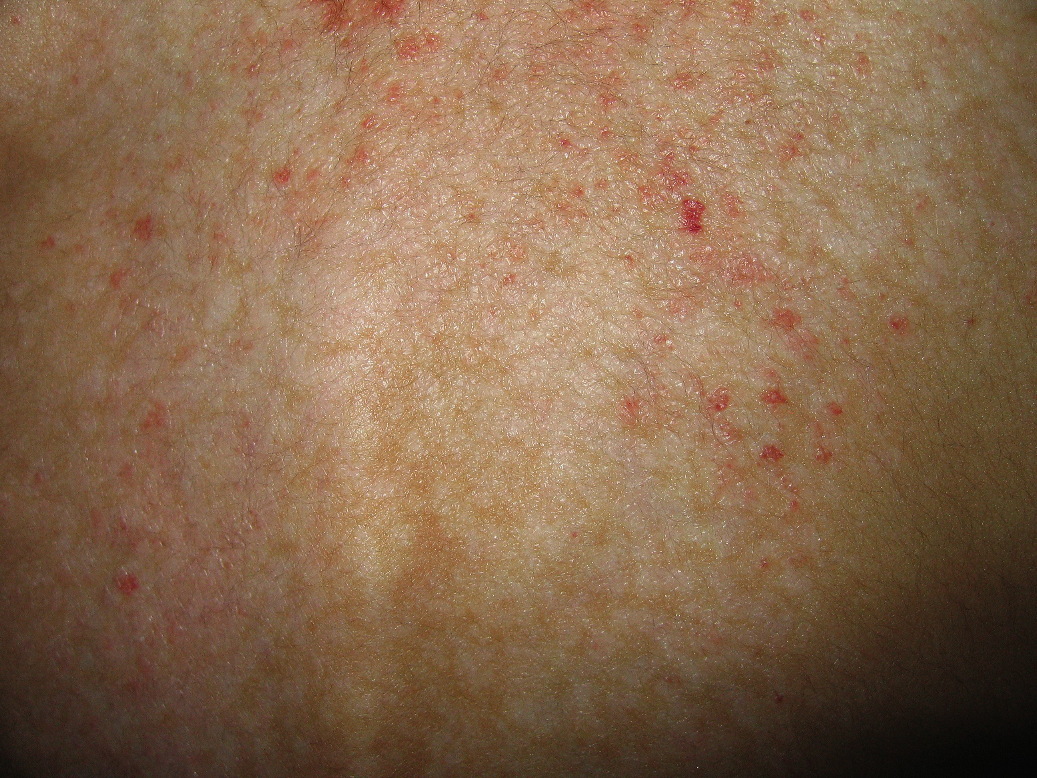
On examination, nearly confluent erythematous macules and papules were present throughout the scalp (Fig. 1) and extended down the midline of his neck and back. Some of the lesions had an overlying yellow crust, whereas others were petechial or purpuric. In addition, hypopigmented macules were noted in areas of partial resolution (Fig. 2). No respiratory distress, lymphadenopathy, or hepatosplenomegaly was appreciated.
 |  |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1: Erythematous crusted plaques and hypopigmented macules on the scalp. Figure 2: Hemorrhagic and hypopigmented macules on back | |
Laboratory testing revealed total bilirubin of 2.3 mg/dl (normal 0.2-1.3), direct bilirubin of 1.2 mg/dl (normal 0.1-0.3), a prothrombin time of 17.6 seconds (normal 12.6-15.2) and serum albumin level of 3.2 g/dl (normal 3.5-4.8). Because the child appeared malnourished, a vitamin deficiency was suspected, but serum folate, vitamin A, vitamin B12, and vitamin D levels were within normal limits. The patient had a normal serum glucose level, and urine osmolality was markedly low at 9 mosm/k (normal 300-900).
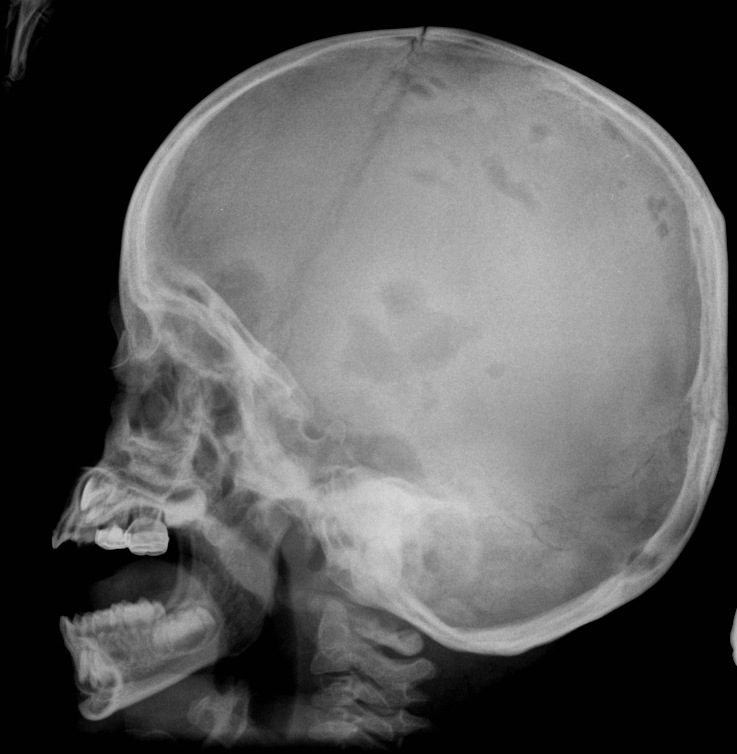
Plane skull x-ray films showed multiple irregular calvarial osteolytic lesions (Fig. 3). A complete skeletal survey additionally revealed osteolytic lesions of the right ileum and superior aspect of the left iliac wing. On MRI a thickened pituitary stalk and absent pituitary gland was noted.
 |  |
| Figure 3 | Figure 4 |
|---|---|
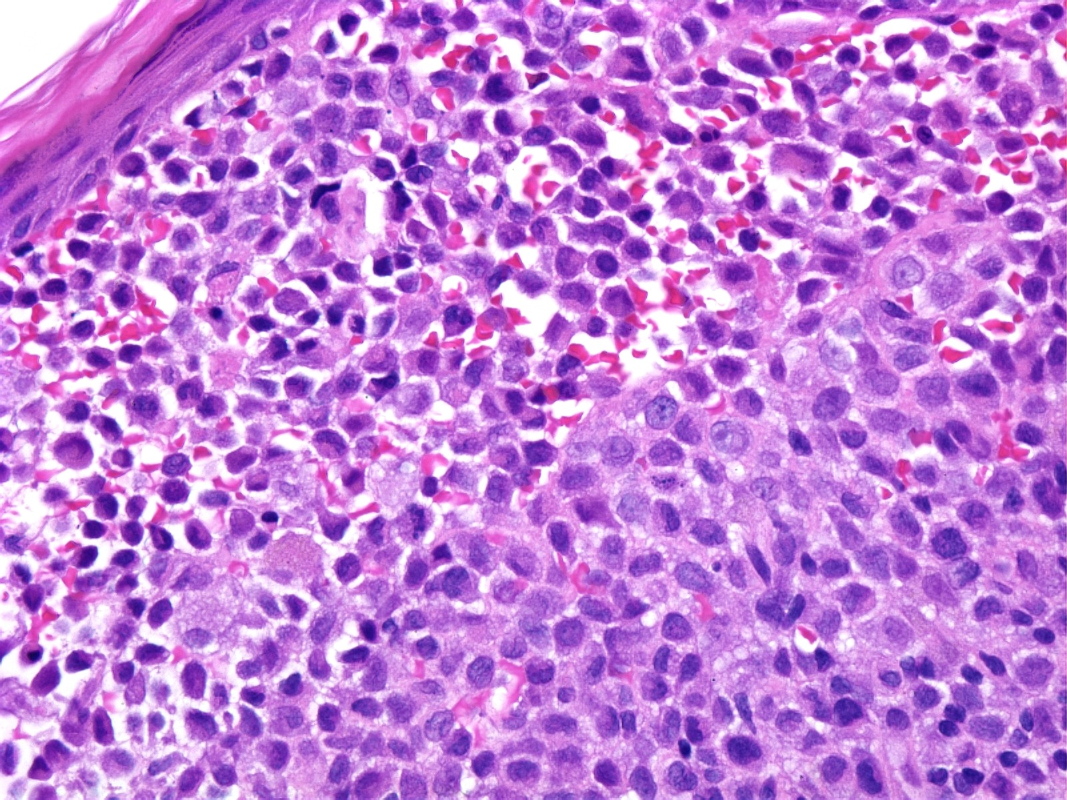
| Figure 3: Osteolytic skull lesions lateral view Figure 4: Diffuse replacement of the superficial dermis by Langerhans Cells (H & E, 10X) | |
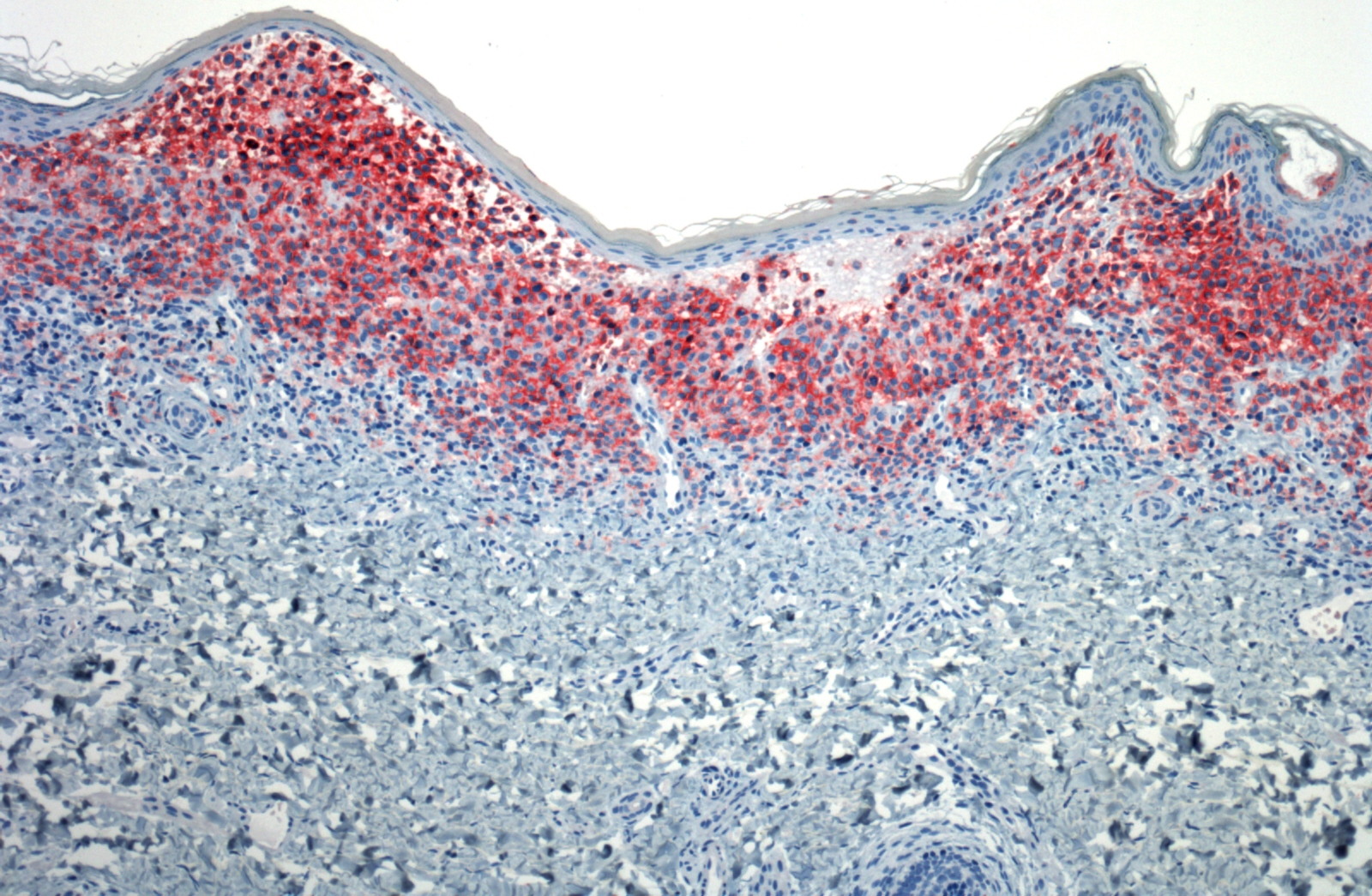 |
| Figure 5 |
|---|
| Figure 5: Marked decoration of Langerhans cells with CD1a (AEC, 10X) |
Histologic examination of a punch biopsy obtained from a lesion on the back revealed diffuse replacement of the superficial dermis by enlarged mononuclear cells admixed with numerous eosinophils. These mononuclear cells had abundant eosinophilic cytoplasm and reniform nuclei. The epidermis was focally ulcerated adjacent to areas in which there was epidermotropism of the mononuclear cells (Fig. 4). Tumoral cells demonstrated strong positivity with S100 and CD1a immunohistochemical stains (Fig. 5). Bone marrow aspirates demonstrated increased cellularity, with a focal increase of eosinophils and abnormal large histiocytes suggestive of LCH; however, the cells of interest were negative with S100 and CD1 immunohistochemical stains.
The patient was diagnosed with LCH with multi-organ involvement and organ failure. The child was enrolled in a Stage IIIb clinical trial sponsored by the Histiocyte Society. In brief, the chemotherapy protocol consisted of 6 weeks of prednisone, intravenous vinblastine, and methotrexate; this was followed by 12 months of maintenance therapy with mercaptopurine, methotrexate, and intravenous pulses of vinblastine and oral prednisolone. Mucositis and neutropenia developed early in the course of his chemotherapy, and during this period, candidal sepsis occurred as a result of colonization of the central line placed for total parental nutrition.
Because of the patient's poor nutritional state and continued emesis, an upper gastrointestinal evaluation with endoscopy was performed. Delayed gastric emptying and poor filling of the duodenum and proximal jejunum was suggestive of duodenal obstruction. Furthermore, mucosal abnormalities of the esophagus and duodenum were appreciated on endoscopic examination. Biopsies taken from these areas revealed mild villous blunting and findings consistent with reflux esophagitis; however, there was no evidence of involvement by LCH.
During the patient's two-month hospitalization his clinical course was further complicated by hypertension and hyperglycemia, both of which necessitated treatment. Twenty-four months after completion of the treatment protocol, he is without evidence of cutaneous or systemic LCH and is taking only desmopressin acetate to control his diabetes insipidus.
Discussion
Clinically and histologically the histiocytoses comprise a diverse group of proliferative disorders characterized by the infiltration and accumulation of histiocytes and other effector cells of the immune system within various tissues. The generic term "histiocyte" refers to several types of cells including: monocytes/macrophages, dermal/interstitial dendritic cells and Langerhans cells (LCs). Histiocytes are critical cells of the immune system: macrophages primarily act as scavenger cells that process antigens whereas LCs serve primarily as antigen presenting cells.
In the past, there had been a great deal of confusion as to how to classify the histiocytoses since the exact ontogeny was not completely understood. However, with the advent of immunohistochemical stains, the Histocyte Society proposed reclassification of these disorders based upon the predominant cell type in the infiltrate. This initial classification system included: Langerhans histiocytosis (Class I), non-Langerhans cell histiocytosis (Class II), and malignant histiocytosis (Class III) [1]. As more information has become available, a revised classification schema was proposed and includes: dendritic cell disorders, macrophage-related disorders, and lastly, malignant histiocytic disorders [2].
One of the more common histiocytoses is LCH, which occurs as a result of a monoclonal proliferation of the pathological LC in a variety of organs, including the skin. Although the cells seen in LCH closely resemble normal LC, they appear more immature (expressing CD14), less dendritic, and more mitotically active (approximately 3-48% cells are in division), but possess reduced to absent antigen-presenting capability [3].
The incidence of LCH ranges from 0.5 to 5.4 cases per million persons per year, depending upon the age of the population investigated [4, 5, 6]. Although the disease can present at any age, it usually presents within the first few years of life and has a slight male predominance. The clinical presentation can range from localized disease, which may spontaneously resolve, to widely disseminated disease with organ failure and death.
Historically, LCH was sub-categorized eponymously based upon the different clinical manifestations. However, with the advent of ultrastructural studies and immunohistochemical stains, conditions that were once believed to be separate entities have been unified under the rubric of LCH. Included in this category, are conditions that were previously designated as eosinophilic granuloma, Hand-Schüller-Christian disease, Letterer-Siwe disease, histiocytosis X, pure cutaneous histiocytosis, congenital self-healing reticulohistiocytosis, Hashimoto and Pritzker disease, Langerhans-cell granulomatosis, type II histiocytosis, and the generic term non-lipid reticuloendotheliosis [1, 2].
Langerhans cell histiocytosis may involve almost any organ system, but the frequency of involvement, as well as the extent of the disease, is often age dependent. Several large retrospective studies consisting of neonates and children under the age of 4 have shown that 51 percent to 71 percent of children with LCH present with multiorgan disease [4, 7, 8, 9, 10]. Although prior studies have shown that adults more frequently present with single system disease, a recent large retrospective study showed that up to 68.9 percent of adults have multisystem involvement [11].
Despite the fact that LCH was first identified at the turn of the 20th century, the etiology remains unknown. There is ongoing debate as to whether LCH represents an abnormal immune response to an unidentified antigen or whether it is truly a neoplastic process [12]. Clonality of the proliferating cells has been cited to support the concept of a neoplastic process; however, clonality does not necessarily imply malignancy and can be seen in benign reactive processes [13, 14]. Although no clear etiology has been identified, the general consensus is that patients with LCH have a dysregulated immune response with failed transition from "innate" to "adaptive" immunity [7, 15]. The LCs in LCH manifest an activated immunophenotype, resulting in their increased proliferation and migration. Aberrant or uncontrolled cytokine production by these inflammatory cells likely results not only in further proliferation of LCs, but also contributes to the pathological sequelae of LCH, including fever, fibrosis, bone resorption, and necrosis [13, 16].
The cornerstone of diagnosis in LCH includes identification of the characteristic clinical features, but also requires corroborating histopathological and immunohistochemical findings [1, 17]. Since 50-80 percent of cases manifest cutaneous involvement, a skin biopsy provides a rapid and accessible means to secure the diagnosis. A presumptive diagnosis of LCH can be made based upon light microscopic findings and a compatible clinical picture; but a definitive diagnosis, requires that the lesional cells exhibit positive staining with S-100 and CD1a [2, 17]. Although CD1a is fairly specific, it also labels cortical thymocytes and interdigitating dendritic cells within the dermis and lymph nodes. Therefore, the sine qua non to identify Birbeck granules is transmission electron microscopy (TEM) [1, 2]. Despite the fact that TEM is the "gold standard," this technique is rarely performed today because immunohistochemical stains have become widely available, easy to use, and relatively inexpensive. Furthermore, the number of LCs with identifiable Birbeck granules can vary dependent upon the type of tissue sampled: limited numbers are visualized in biopsies taken from lesional tissue from the liver, spleen, gastrointestinal tract and central nervous system [6]. Therefore, other pathognomonic surface markers are being sought. Langerin (CD 207) is a relative new monoclonal antibody directed against a type II transmembrane protein associated with Birbeck granules. It appears to be more sensitive and specific for LCs than CD1a. In the future it may be a key component of an immunocytochemical panel to identify LC [3, 16, 18, 19].
The Histiocyte Society has established a set of guidelines to assist in the diagnosis and study of LCH [17]. The initial evaluation consists of a complete physical examination, inclusive of height and weight measurements, in addition to laboratory studies including hematological assays and coagulation studies, liver function tests, and urine osmolality. Although some authorities advocate bone marrow examination in every baseline examination, it is not required unless symptoms or blood tests suggest involvement. Lastly, the patient must have a complete skeletal radiographic survey and chest radiography. Patients with identified abnormalities require more specific studies, such as pulmonary function tests and lung biopsy, small bowel series, liver biopsy, panoramic dental films, CT or MRI of the brain with particular attention paid to the hypothalamic-pituitary axis, endocrine evaluation and otolaryngology consultation with audiogram. Since patients with LCH often have chronic and recurrent disease, follow-up studies are required every month to 6-months, depending upon organ system involvement and the degree of organ dysfunction [17].
To aptly determine a patient's prognosis and treatment protocol, it is currently recommended that patients are risk-stratified based upon the number of organs involved and degree of organ dysfunction [17, 19, 20, 21, 22]. Patients diagnosed with organ dysfunction are further stratified based upon which organ system is involved. Patients with involvement of the spleen, lung, liver or hematopoietic system often have a worse prognosis [12, 14, 20, 22]. Although some studies found that the younger the patient is at the time of diagnosis, the worse the prognosis, this only holds true when multiorgan involvement is present, because neonates who present with isolated cutaneous lesions do exceptionally well [5, 10, 22, 23, 24].
In the past, treatment for LCH was anecdotal and without concensus. However, within the last 20 years several multi-center, randomized therapeutic trials have contributed to a more uniform treatment approach. Patients with limited cutaneous disease typically require no therapy. If therapy is required, topical steroids may be tried as a first line treatment. Patients, who are non-responsive to steroids, can utilize topical nitrogen mustard or PUVA therapy as viable second-line options [10, 25, 26]. For patients who have localized bone lesions, curettage is generally sufficient for diagnosis as well as therapy, although some cases may require intralesional steroids or low dose radiation [27]. Treatment of multi-organ disease is controversial, with some advocating high-dose prednisone as the first-line therapy, whereas others suggesting use of single-agent chemotherapy [25, 28, 29]. More recently, several large cooperative studies have shown that multi-agent chemotherapy that is sustained over a longer duration results in a greater response with fewer reoccurrences [21, 22, 26]. Currently, the LCHIII treatment protocol is probably the most common therapeutic strategy used in children with multiorgan involvement. In the future, monoclonal antibodies that target CD1a or CD207 or specific cytokine inhibitors maybe employed [25, 26].
As a result of recent therapeutic trials, it has been shown that the single best prognostic indicator is a patient's response to chemotherapy during the 6-week induction phase [22, 30-35]. Patients who respond to chemotherapy during the first 6 weeks of therapy have an 88-91 percent survival rate, but survival rates drop to 17-34 percent in patients who fail to respond. It has been advocated that non-responders be identified early so that more aggressive therapy may be employed [22, 26, 30, 35].
Disease activity may be assessed utilizing criteria established by the Histiocyte Society. Under this system, patients are placed into one of four subcategories: non-active disease, evidence of disease with regression, stable disease, and progressive disease. Although this system appears simple, a more objective assessment of disease activity has been recently proposed [36]. The later methodology is based upon obtaining a score derived from three primary parameters including: clinical examination, laboratory evaluation, and radiological findings. Patients with more abnormal findings receive a higher score, and therefore are at greater risk for disease progression and demise.
Despite adequate treatment, it has been shown that survivors of LCH have long-term sequelae, some of which may not become apparent until many years later [37]. One retrospective study analyzed patients who had been treated for multi-organ system LCH and found that 75 percent had detectable long-term sequelae, such as hypothalamic-pituitary dysfunction (50%), cognitive dysfunction (20%), and cerebellar involvement (17.5%) [38]. Some sequelae are multifactorial. For example, growth retardation can occur as a result of gastrointestinal involvement by LCH thereby resulting in malabsorption, or due to deficiencies in growth hormone from anterior pituitary lesions, or from the residua of chemotherapy. Furthermore, long-term complications are not limited to patients with multi-organ system disease and approximately 25 percent of patients with single-system disease experience permanent sequelae [39]. Even patients diagnosed with congenital self-healing LCH have been shown to have late relapses and/or progression to systemic disease [24]. Consequently, all patients with LCH require long-term follow-up to identify disease recurrence or late-stage complications [9]. Lastly, caregivers should be cognizant that patients with LCH are at risk for second malignancies, including solid tumors and hematopoietic conditions.
Conclusion
In summary, LCH represents a disease with a diverse spectrum of clinical manifestations. Herein through this pedagogic case, we summarize the current recommendations of the Histiocyte Society regarding the classification, evaluation, prognosis, and treatment of LCH. A definitive diagnosis of LCH can be made by obtaining a biopsy that yields cells that are morphologically and immunohistochemically compatible with LCs. Proposals by the Histiocyte Society have assisted in the unification of LCH into one disease category and has allowed for more efficient treatment protocols to be designed via risk stratification. The prognosis depends chiefly upon the involvement of multiple organ systems, organ dysfunction and the patient's response to chemotherapy during the initial 6 weeks of treatment.
Acknowledgments: The authors thank the Histiocyte Society for providing protocol information.
References
1. Chu T, D'Angio GJ, Favara B, Ladisch S, Nesbit M, Pritchard J. Histiocytosis Syndromes in Children. Writing Group of the Histiocyte Society. Lancet 1987; 2(8549):41-42. PubMed2. Favara BE, Feller AC, Pauli M, Jaffe ES, Weiss LM, Arico M, Bucsky P, Egeler RM, Elinder G, Gadner H, Gresik M, Henter JI, Imashuku S, Janka-Schaub G, Jaffe R, Ladisch S, Nezelof C, Pritchard J. Contemporary Classification of Histiocytic Disorders. The WHO Committee on Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocytic Society. Med Pediatr Oncol 1997;29(3):157-166. PubMed
3. Geissmann F, Lepelletier Y, Fraitag S, Valladeau J, Bodemer C, Debré M, Leborgne M, Saeland S, Brousse N. Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood 2001;97(5):1241-1248. PubMed
4. [no author listed]. A multicenter retrospective survey of Langerhans' cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans' Cell Study Group. Arch Dis Child 1996;75(1):17-24. PubMed
5. Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer 2007 48(5):555-560. PubMed
6. Schmitz L, Favara BE. Nosology and pathology of Langerhans cell histiocytosis. Hematol Clin North Am 1998;12(2): 222-246. PubMed
7. Bhatia S, Nesbit ME Jr, Egeler RM, Buckley JD, Mertens A, Robison LL. Epidemiologic study of Langerhans cell histiocytosis in children. J Pediatr 1997;130(5):774-784. PubMed
8. Isaacs H Jr. Fetal and neonatal histiocytoses. Pediatr Blood & Cancer 2006; 47(2):123-129. PubMed
9. Longaker, MA, Frieden IJ, LeBoit PE, Sherertz EF. Congenital "self-healing" Langerhans cell histiocytosis: The need for long-term follow-up. J Am Acad Dermatol 1994;31(5 Pt 2):910-916. PubMed
10. Stein SL, Paller AS, Haut PR, Mancini AJ. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med 2001;155(7):778-83. PubMed
11. Arico M, Girschikofsky M, Généreau T, Klersy C, McClain K, Grois N, Emile JF, Lukina E, De Juli E, Danesino C. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocytic Society. Eur J Cancer 2003; 39(16):2341-2348. PubMed
12. Goltzbecker MP, Carpentieri DF, Dormans JP. Langerhans cell histiocytosis: Clinical presentation, pathogenesis, and treatment from the LCH Etiology Research Group at the Children's Hospital of Philadelphia. The University of Pennsylvania Orthopedic Journal 2002;15:67-73.
13. Willman CL, McClain KL. An update on clonality, cytokines, and a viral etiology in Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998;12(2):408-416. PubMed
14. Egeler RM, D'Angio GJ. Langerhans cell histiocytosis. J Pediatr 1995;127(1):1-11. PubMed
15. Savasan, S. An enigmatic disease: childhood Langerhans cell histiocytosis in 2005. Int J Dermatol 2006;45(3):182-188. PubMed
16. Bechan GI, Egeler RM, Arceci RJ. Biology of Langerhans cells and Langerhans cell histiocytosis. Int Rev Cytol 2006;254:1-43. PubMed
17. Broadbent V, Gadner H, Komp DM, Ladisch S: Histiocytosis syndromes in children: II. Approach to the clinical and laboratory evaluation of children with Langerhans cell histiocytosis. Clinical Writing Group of the Histiocyte Society. Med Pediatr Oncol 1989;17(6):492-5. PubMed
18. Nezelof C, Basset F. Langerhans cell histiocytosis research. Past, present, and future. Hematol Oncol Clin North Am 1998;12(2):385-406. PubMed
19. Hicks J, Fliatz M. Langerhans cell histiocytosis: Current insights in a molecular age with emphasis on clinical oral and maxillofacial pathology practice. Oral Surg Oral Med Oral Pathol Pral Radiol Endod 2005;100(2 suppl):S42-S66. PubMed
20. Lahey E. Histiocytosis-X: an analysis of prognostic factors. J Pediatr 1975; 87(2):184-189. PubMed
21. Arceci RJ, Brenner MK, Pritchard J. Controversies and new approaches to treatment of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998;12(2):339-357. PubMed
22. Gadner H, Grois N, Arico M, Broadbent V, Ceci A, Jakobson A, Komp D, Nicholson S, Potschger U, Pritchard J, Ladisch S; Histocyte Society. A randomized trial of treatment for multisystem Langerhans cell histiocytosis. J Pediatr 2001;138 (5):728-734. PubMed
23. Rivera-Luna R, Martinez-Guerra G, Altamirano-Alverez E, Martinez-Avalos A, Cardenas-Cardoz R, Ayon-Cardenas A, Ruiz-Maldonado R, Lopez-Corella E. Langerhans cell histiocytosis: Clinical experience with 124 patients. Pediatr Dermatol 1998;5(3):145-150.PubMed
24. Esterly NB, Mauer HS, Gonzalez-Crussi F. Histiocytosis X: A Seven-Year Experience at a Children's Hospital. J Am Acad Dermatol 1985;13(3):481-496. PubMed
25. Arico M, Egeler RM. Clinical aspects of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998;12(2):247-259. PubMed
26. Broadbent V, Gadner H. Current therapy for Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998;12(2):327-338. PubMed
27. Kilpatrick SE, Wenger DE, Gilchrist GS, Shives TC, Wollan PC, Unni KK. Langerhans cell histiocytosis (Histiocytosis X) of bone. A clinicopathologic analysis of 263 pediatric and adult cases. Cancer 1995;76(12): 2471-2484. PubMed
28. Howarth DM, Gilchrist GS, Mullan BP, Wiseman GA, Edmonson JH, Schomberg PJ. Langerhans cell histiocytosis: diagnosis, natural history, management, and outcome. Cancer 1999;85(10):2278-90. PubMed
29. McLelland J, Broadbent V, Yeomans E, Malone, Pritchard J. Langerhans cell histiocytosis: the case for conservative treatment. Arch of Disease Child 1990; 65(3):301-303. PubMed
30. Ladisch S, Gadner H, Arico M, Broadbent V, Grois N, Jakobson A, Komp D, Nicholson HS. LCH-I: a randomized trial of etoposide verses vinblastine in disseminated Langerhans cell histiocytosis. The Histiocyte Society. Med Pediatr Oncol 1994;23(2):107-110. PubMed
31. Gadner H, Heitger A, Grois N, Gatterer-Menz I, Ladisch S. Treatment strategy for disseminated Langerhans cell histiocytosis. Med Pediatr Oncol 1994;23(2):72-80. PubMed
32. Minkov M, Grois N, Heitger A, Potschger U, Westermeier T, Gadner H. Treatment of multisystem Langerhans cell histiocytosis. Results of DAL-HX 83 and DAL-HL 90 studies. DAL-HX Study Group. Klin Pediatr 2000;212(4):139-144. PubMed
33. Ceci A, de Terlizzi M, Colella R, Loiacono G, Balducci D, Surcio G, Castello M, Testi AM, De Bernardi B, Indolfi P et al. Langerhans cell histiocytosis in childhood: results from the Italian cooperative AIEOP-CNR-HX'83 Study. Med Pediatr Oncol 1993;21(4):259-264. PubMed
34. McClain KL, Kozinetz CA. A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr Blood Cancer 2007;48(1):44-49. PubMed
35. Mottl H, Ganevovà Radvanská J, Cháňová M, Šmelhaus V, Kodet R, Mrhalová, Tichý M. Outcome of treatment of Langerhans cell histiocytosis to the LCH II protocol. Čas LéK Česk 2005;144(11):753-755. PubMed
36. Donadieu J, Piguet C, Bernard F, Barkaoui M, Ouache M, Bertrand Y, Ibrahim H, Emile JF, Hermine O, Tazi A, Genereau T, Thomas C. A New clinical score for disease activity in Langerhans cell histiocytosis. Pediatric Blood & Cancer 2004;43(7):770-776. PubMed
37. Haupt R, Naduri V, Calevo MG, Bernstrand C, Braier JL, Broadbent V, Rey G, McClain KL, Janka-Schaub G, Egeler RM. Permanent Consequences in Langerhans cell histiocytosis patients: A pilot study from the Histiocyte Society-Late effects study group. Pediatr Blood Cancer 2004;42(5):438-444. PubMed
38. Nanduri VR, Pritchard J, Levitt G, Glaser AW. Long-term morbidity and health related quality of life after multi-system Langerhans cell histiocytosis. Eur J Cancer 2006;42(15):2563-2569. PubMed
39. Titigemeyer C, Grois N, Minkov M, Flucher-Wolfram B, Gatterer-Menz I, Gadner. Pattern and course of single system disease in Langerhans cell histiocytosis data from the DAL-HX83 and 90 studies. Med Pediatr Oncol 2001;37(2):108-14. PubMed
© 2008 Dermatology Online Journal