Waardenburg Syndrome type 1: A case report
Published Web Location
https://doi.org/10.5070/D314k128r2Main Content
Waardenburg Syndrome type 1: A case report
Gulsen Tukenmez Demirci MD, Guldehan Atıs MD, Ilknur Kıvanc Altunay MD
Dermatology Online Journal 17 (11): 3
Şişli Etfal Training and Research Hospital, Istanbul, TurkeyAbstract
Waardenburg Syndrome (WS) is a rare hereditary disorder that is characterized by the clinical manifestations of oculocutaneous anomalies of pigmentation, congenital deafness, dystopia canthorum, and broad nasal root. It demonstrates both genetically and clinically heterogenous characteristics. In this article, we report an 11-month-old boy with WS1, one of four clinicat types of WS. He exhibited white forelock, hypopigmented macules and patches, heterochromia irides, and dystopia canthorum.
Introduction
Waardenburg Syndrome (WS) is a rare autosomal dominant or autosomal recessive genetic disorder characterized by achromia of the hair, skin (or both), congenital deafness, partial or total heterochromia iridis (including isohypochromia), hypertrichosis of the medial part of the eyebrows (synophrys), broad and high nasal root, and dystopia canthorum (increase in the distance between the inner canthi with normal inter-pupillary distance) [1]. Waardenburg Syndrome is classified into four clinical types according to the presence of variable clinical characteristics and additional signs, as WS 1, WS 2, WS 3, and WS 4 [2, 3, 4]. Diagnostic criteria for WS have been proposed by the Waardenburg Consortium (Farrer et al 1992). See Table 1. Two major or one major plus two minor criteria are necessary for the diagnosis of WS1. WS2 lacks dystopia canthorum. WS3 is quite similiar to WS1 but it is associated with upper limb defects. WS4 is associated with Hirschsprung disease [5, 6].
We report, herein, an 11-month-old boy with abnormal pigmentation of the hair, skin, and iris, along with dystopia canthorum. He was classified as WS1 because of the absence of sensorineural deafness.
Case report
 |  |
| Figure 1 | Figure 2 |
|---|---|
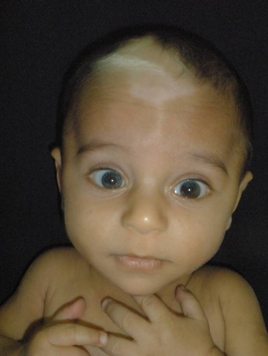
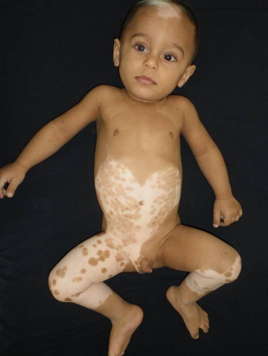
| Figure 1. Triangular hypopigmented patch and white forelock on the frontal region Figure 2. A large achromic patch with a sharp but irregular border, which has pigmented, partly grouped, macules in various sizes on the abdomen. | |
 |
| Figure 3 |
|---|
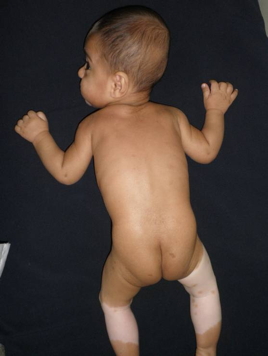
| Figure 3. A few hyperpigmented macules are conspicuous on the normal skin of gluteal region. |
An 11-month-old boy presented to our clinic with white forelock on the anterior scalp and hypopigmented macules on the trunk, all present since birth. He was healthy except for his skin condition. Delivery was normal and he was born at term. Family history was unremarkable. No clinical abnormality or skin disease was present in his parents, but they were first degree, consanguineous. Dermatological examination showed a triangular hypopigmented patch and white forelock on the frontal scalp and forehead region (Figure 1). A large achromic patch with sharp, but irregular border, which had partly grouped, pigmented macules in various sizes was noted on the abdomen and also on the right thigh and left leg. A few hyperpigmented macules were conspicuous on the normal skin of the gluteal region, as well (Figures 2 and 3). Ophthalmological examination demonstrated iris heterochromia and dystopia canthorum. However, a neurological examination revealed no abnormal findings. Cranial magnetic resonance revealed no abnormality. The Bera Test was performed to evaluate sensorineural deafness and it was within normal limits. Laboratory investigations were within normal limits. The patient was diagnosed as WS1 with these findings.
Discussion
The clinical variability of WS is attributed to the different penetrance and expression of the responsible genes, just as in other genetic syndromes. WS1 is usually diagnosed by clinical findings that include sensorineural hearing loss, pigmentary changes in the hair and eyes, and dystopia canthorum. Dystopia canthorum is a crucial clinical finding that mostly distinguishes WS1 from WS2 [7]. Dystopia canthorum is defined as prominent broad nasal root with increased intercanthal distance, but it is different than ocular hypertelorism. Dystopia canthorum is identified by calculation of the W index. The measurements necessary to calculate the W index (in mm) are as follows: inner canthal distance (a), interpupillary distance (b), and outer canthal distance (c) [8].
- Calculate X = (2a – (0.2119c + 3.909))/c
- Calculate Y = (2a – (0.2479b + 3.909))/b
- Calculate W = X + Y + a/b
An abnormal W index result is greater than 1.95. Previously, a W index of greater than 2.07 was necessary to diagnose WS1 in an individual meeting all of the other diagnostic criteria. With molecular analysis, a family previously classified clinically as having WS2 based on the W index was found to have a PAX3 mutation and was reclassified as having WS1 [9]. Hence, the W index threshold was reduced to its current value of greater than 1.95 [7].
If the average W index in a family is less than 1.95, the diagnosis is WS2. Sensorineural hearing loss and heterochromia iridis are the two most characteristic features of WS2. Both are more common in WS2 than in WS1. White forelock and leukoderma are both more common in WS1 than in WS2 [10].
Our case was diagnosed as WS1 with 3 major and 2 minor criteria. Diagnostic criteria have been identified in the Waardenburg consortium with the presence of white forelock, heterochromia iridis, and dystopia canthorum. The characteristic clinical features in differentiating WS1 from other WS types are summarized in Table 2. Cutaneous pigmentary defects occur in 8.3-50 percent of WS patients. Piebaldism-like achromic spots may occur and hyperpigmented macules may also be seen. Partial or complete heterochromia iridis occurs in 21-28 percent of patients with WS [5]. Our case had all these clinical findings.
Hearing loss, which is one of the major criteria of the WS consortium, was not present in our patient. WS is reported to account for 2 percent of congenital deafness [11]. This clinical finding is not a universal feature of WS, but penetrance of sensorineural hearing loss has been observed to be 69 percent in WS1 and 87 percent in WS2 after excluding probands ascertained through their hearing loss [5]. Whereas pigmentation abnormalities do not herald any survival problem, hearing loss seems to be the most important prognostic factor because of impairment of life quality and poor cognitive abilities in WS1. The earlier it is diagnosed, cochlear implantation will more easily lead to improvement of speech discrimination and spoken language [12].
Multiple genes have been implicated in the syndrome. Abnormalities in the PAX3 gene (paired box gene 3) accounts for most of WS1 and WS3 patients. MITF (microphthalmia associated transcription factor) gene abnormality is responsible for WS2. WS4 is heterogeneous, with reported mutations in EDN3 (endothelin 3), in its receptor EDNRB (endothelin receptor type B), or in SOX10 (SRY-sex determining region Y-box 10) [11].
The PAX3 gene on chromosome 2q35 is a transcription factor that plays a major role in embryogenesis [13]. WS1 is inherited in an autosomal dominant manner. The majority of probands have an affected parent, whereas a minority of probands do not have an affected parent ane are presumed to have a de novo mutation [7]. We could not perform the PAX3 gene sequence and deletion/duplication analysis in our case, but it seems to be a de novo mutation because the patient has healthy parents and family members.
References
1. Ortonne JP. Vitiligo and other disorders of hypopigmentation. In Bolognia JL, Jorizzo JL, Rapini RP et al eds. Dermatology. 2nd edn. Spain: mosby elsevier, 2008:924-9252. Soni C.R, Kumar G. Child neurology: A patient with dissimilar eye color and deafness. Neurology 2010;74:25-6. [PubMed]
3. Qin W, Shu A, Qian X, Gao J, Xing Q, Zhang J, Zheng Y, Li X, Li S, Feng G, He L. A novel mutation of PAX3 in Chinese family with Waardenburg syndrome. Mol Vis. 2006;12:1001-8. [PubMed]
4. Tomita Y, Suzuki T. Genetics of pigmentary disorder. Am J Med Genet C Semin Med Genet. 2004 ;15:75-81. [PubMed]
5. Tagra S, Talwar A.K, Walia R.L.S, Sidhu P. Waardenburg syndrome. Indian J Dermato Venereol Leprol. 2006 Jul-Aug;72(4):326. [PubMed]
6. Arca E, Özkan İ, Taştan H.B, Gür A.R.İki Waardenburg Sendromu Olgusu. Turkderm. 2006;40:64-67
7. Milunsky JM. Waardenburg syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of University of Washington, Seattle; 1993-2001 Jul 30 [updated 2009 Aug 4]. [PubMed]
8. Arias S, Mota M. Apparent non-penetrance for dystopia in Waardenburg syndrome type I, with some hints on the diagnosis of dystopia canthorum. J Genet Hum. 1978;26:103-131.
9. Tassabehji M, Read AP, Newton VE, Patton M, Gruss P, Harris R, Strachan T. Mutations in the PAX3 gene causing Waardenburg syndrome type 1 and type 2. Nat Genet. 1993;3:26-30.
10. Liu XZ, Newton VE, Read AP. Waardenburg syndrome type II: phenotypic findings and diagnostic criteria. Am J Med Genet. 1995;55:95-100
11. Yang S, Cao J, Zhang R, Liu L, liu X, Zhang X, Kang D, Li M, Han D, Yuan H, YangW. Nonsense mutations in the PAX3 gene cause Waardenburg syndrome type 1 in two Chinese patients. Chin Med J. 2007;120(1):46-49. [PubMed]
12. Deka RC, Sika K, Chaturvedy G, Singh CA, Venkat Karthikeyan C, Kumar R, Agarwal S. Cochlear implantation in Waardenburg syndrome: The Indian scenario. Acta Otolaryngol. 2010;130(10):1097-100 [PubMed]
13. Eigelshoven S, Kameda G, Kortüm AK, Hübsch S, Angerstein W, Singh P, Vöhringer R, Goecke T, Mayatepek E, Ruzicka T, Wildhardt G, Meissner T, Kruse R Waardenburg syndrome type I with heterochromia iridis and circumscribed hypopigmentation of the skin. Pediatr Dermatol. 2009 Nov-Dec;26(6):759-61. [PubMed]
© 2011 Dermatology Online Journal