Keratoderma hereditarium mutilans (Vohwinkel syndrome) in three siblings
Published Web Location
https://doi.org/10.5070/D31452s9p1Main Content
Keratoderma hereditarium mutilans (Vohwinkel syndrome) in three siblings
Arfan ul Bari
Dermatology Online Journal 12 (7): 10
Combined Military Hospital, Muzaffarabad, AJK, Pakistan. albariul@yahoo.com
Abstract
Vohwinkel syndrome or keratoderma hereditaria mutilans is a rare diffuse, honeycombed, palmoplantar keratosis usually accompanied by pseudoainhum near the distal interphalangeal creases. The syndrome is reported in three siblings (two brothers and one sister) out of five, who had been having this problem since early childhood.
Hereditary palmoplantar keratodermas (PPKs) are a highly heterogeneous group of skin diseases. They can be inherited in both an autosomal dominant and recessive fashions. They are classified by morphology and distribution of thickening, genetic transmission, presence of skin lesions on areas other than palms and soles and age of onset [1]. The elucidation of the underlying gene defects for many of the PPKs further aids in their classification [2]. Simple keratodermas manifest as lesions only on the palmoplantar skin, whereas complex keratodermas are associated with lesions of non-volar skin, hair, teeth, nails or sweat glands. Syndromic keratodermas are associated with abnormalities of other organs such as deafness, cancer, cardiomyopathy, and adrenal insufficiency. Simple keratodermas are further divided into three main groups, the diffuse, the focal and the punctate PPKs [1, 2, 3].
Vohwinkel syndrome, known as keratoderma hereditaria mutilans, is a syndromic form of diffuse PPK that manifests as hyperkeratosis of the palms and soles with a honeycomb appearance. It is a rare autosomal dominant PPK, which was first described in 1929 by Vohvinkel [4]. Clinically, this condition manifests in infants, becomes transgredient during childhood, and constricting fibrous bands appear at later stages on the digits and can lead to progressive strangulation and autoamputation (pseudoainhum). Starfish-shaped keratoses may occur on the elbows, knees and the dorsa of the hands and feet, a characteristic feature of this disorder. Some associated features have been noted and include alopecia, hearing loss, spastic paraplegia, myopathy, ichthyosiform dermatoses, and nail abnormalities [1, 3, 5]. Histopathologically, the skin in Vohwinkel syndrome shows hyperkeratosis with parakeratosis, acanthosis and hypergranulosis. There is some overlap of the clinical features of Vohwinkel syndrome with those of other disorders. For example, pseudoainhum can be seen in discoid lupus erythematosus and in mal de Meleda [6, 7]. Recent molecular biological studies indicate the presence of two variants of Vohwinkel syndrome, an (1) ichthyosis-associated variant, associated with an insertional mutation of the loricrin gene, and a (2) deafness-associated variant, associated with a missense mutation of the connexin-26 gene [3, 8, 9, 10].
The treatment of all types of hereditary keratoderma is difficult. It tends to be symptomatic and may vary from simple measures, such as saltwater soaks and paring, to topical keratolytics, systemic retinoids, or reconstructive surgery with total excision of the hyperkeratotic skin followed by grafting. The mainstays of treatment include topical keratolytics, topical retinoids and oral retinoids. Oral retinoids are effective, especially in some hereditary PPKs [10]. The prognosis is good as long as medications are used; patients with this syndrome may have a normal life span.
Clinical synopsis
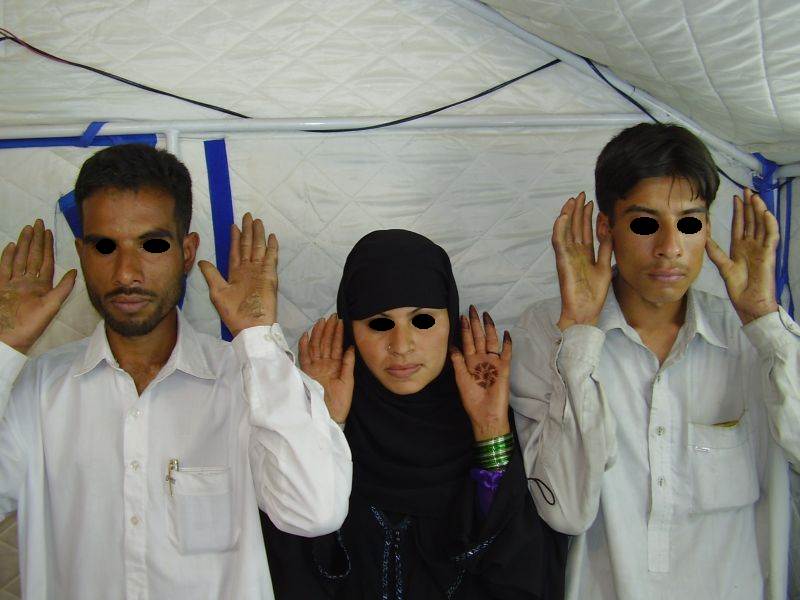
|

|
| Figure 1a | Figure 1b |
|---|---|
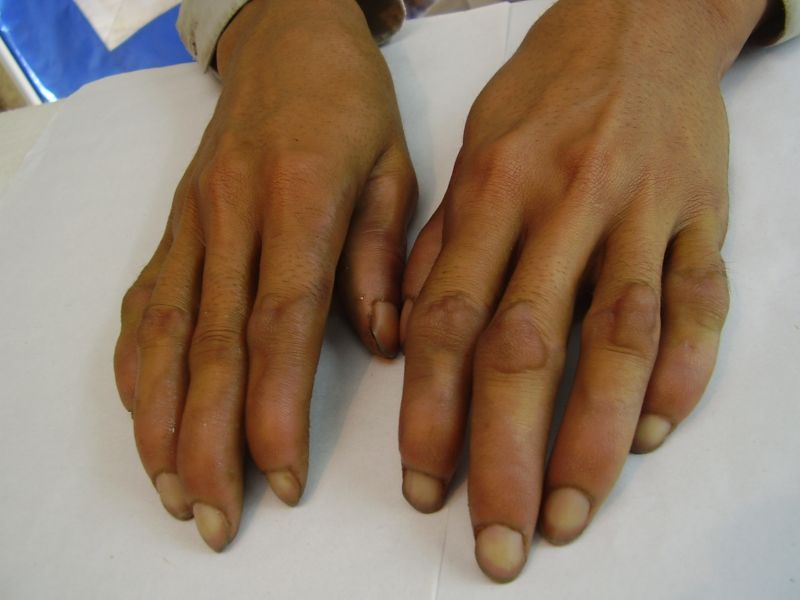
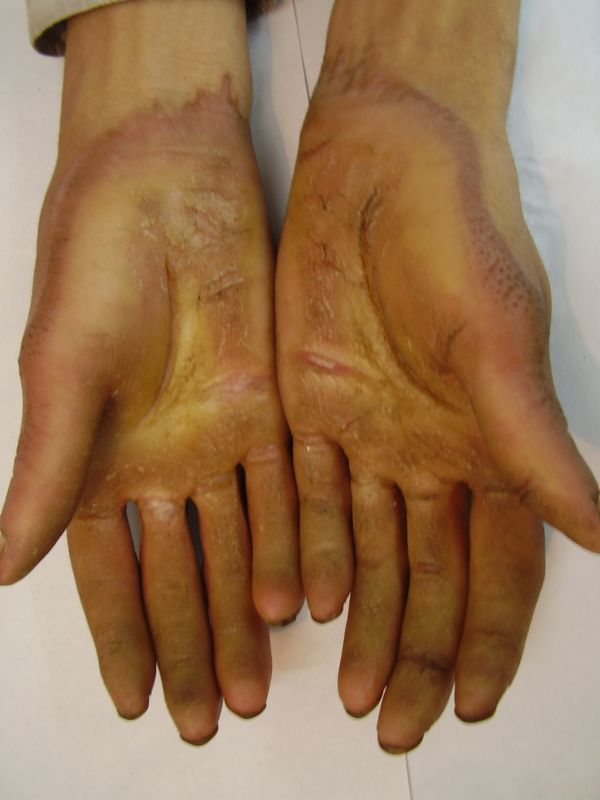
| Three siblings (two brothers and one sister)(1a) showing diffuse keratoderma over palms (1b) and figurate hyperkeratotic plaques over dorsal aspects of hands (1c) | |
|
| Figure 1c |
|---|
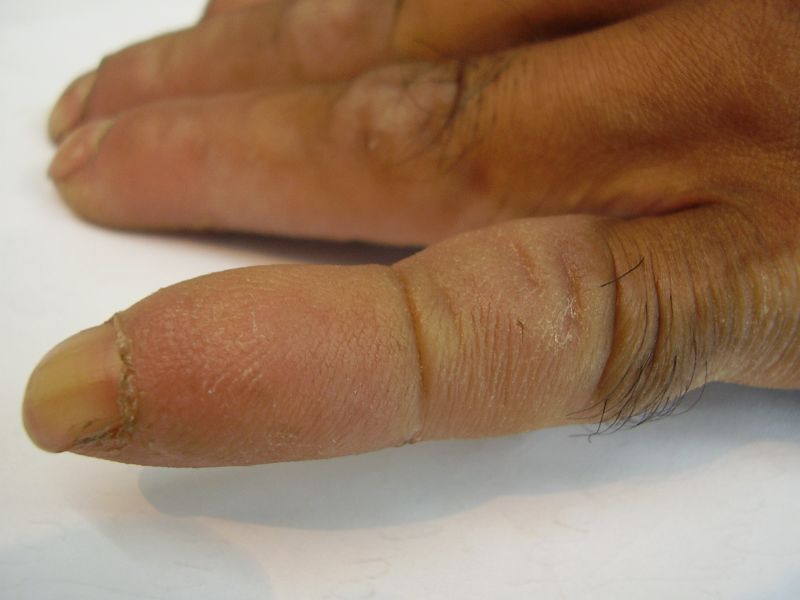
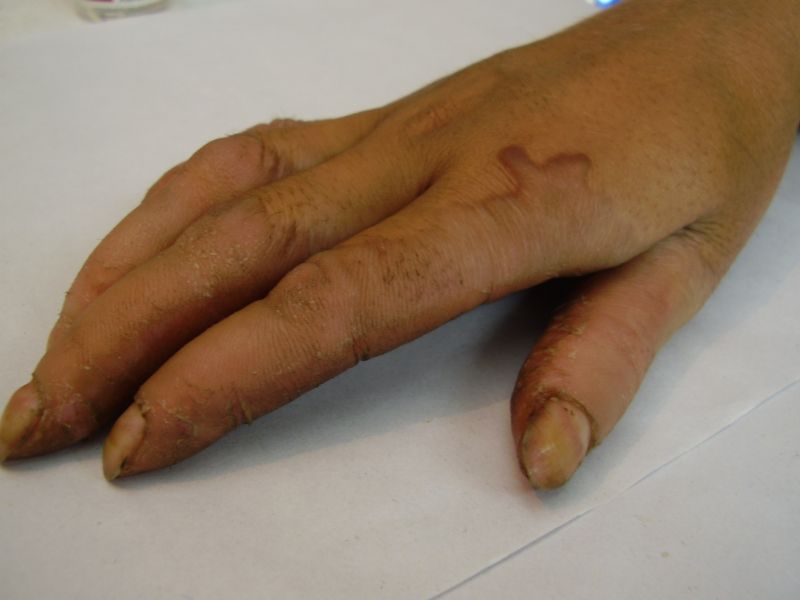
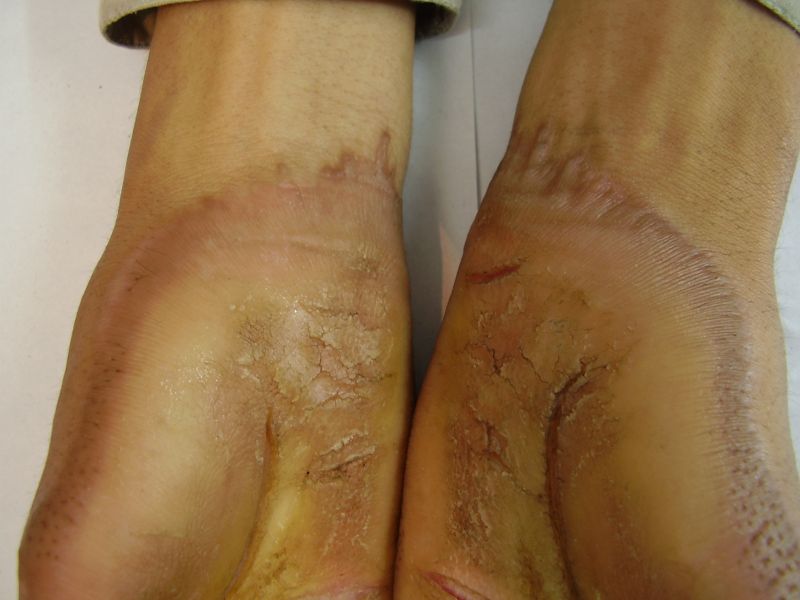
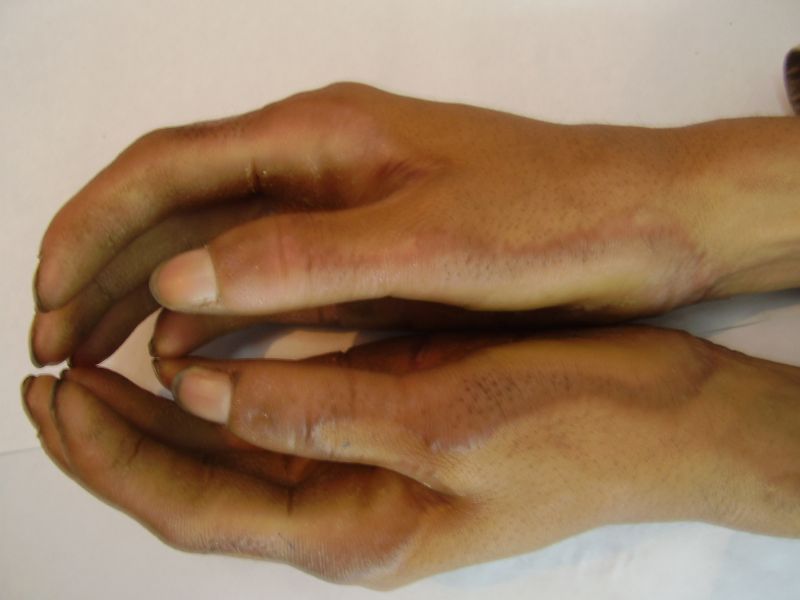
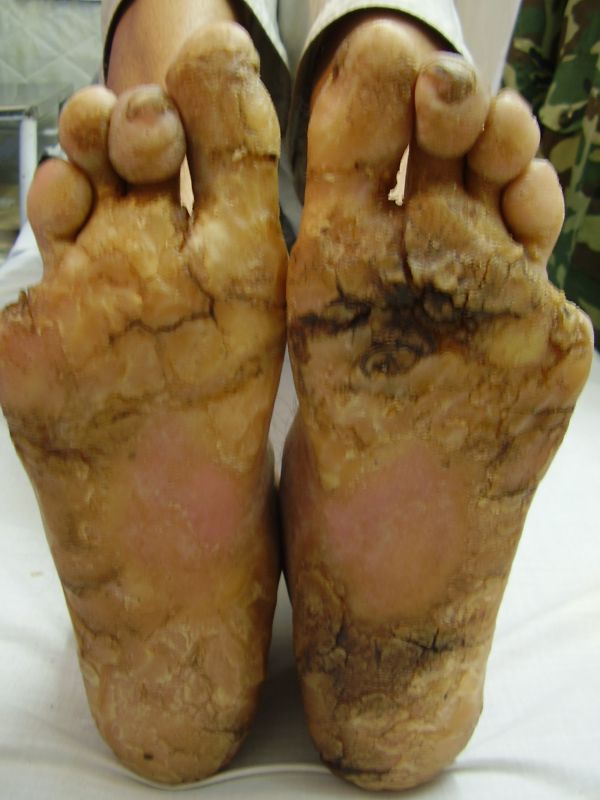
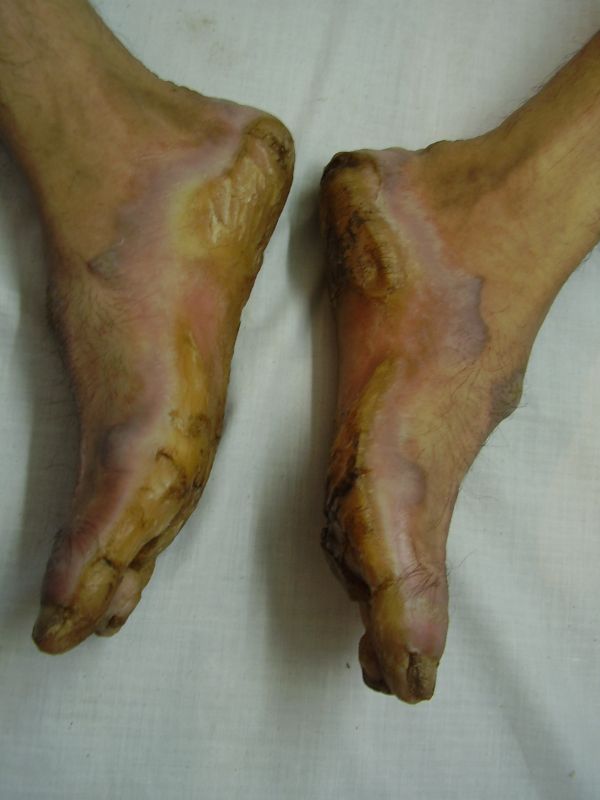
Three adult siblings, aged 23, 21 and 18 years (two brothers and one sister) (Fig. 1a) presented to the Dermatology outpatient clinic of the Combined Military Hospital, Muzaffarabad, Pakistan with markedly thickened and cracked palms and soles since their early childhood. They were born of a non-consanguineous marriage and were apparently normal at birth with normal milestones. The parents first noted thickening of the skin of the soles at the age of 2 years. Soon thereafter, the process affected the palms too. This thickening gradually extended to the dorsal surfaces of the hands and feet with a well-defined reddish thick margin. Meanwhile some isolated thick plaques also appeared on the dorsum of the hands and feet. At the age of 15, constriction bands appeared in one patient (the younger brother) around the proximal interphalangeal joints of both fifth toes resulting in subsequent painful autoamputation of the digits in 2 years (Fig. 4f). A similar constriction band involving the fifth finger of the left hand has also developed in his elder brother (Fig. 2d). Thickening of the soles was more progressive and at times caused restriction of movement. All patients could do some manual work only after applying emollient to their palms. Stiffness of the hands and feet tends to worsen in the winter months and during the hot humid season the palms and soles become foul-smelling at times (possibly because of hyperhidrosis and secondary bacterial and fungal infections). The patients have been applying various keratolytics combined with emollients with transient partial relief of the symptoms. None of them had any history of deafness or ichthyotic skin and there was no family history of similar illness.
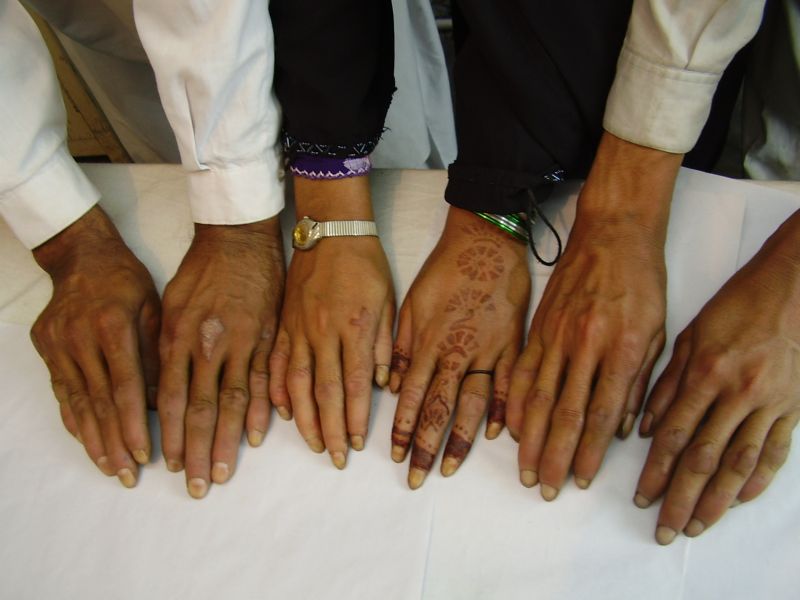
|
|
| Figure 2a | Figure 2b |
|---|
|
|
| Figure 2c | Figure 2d |
|---|
|
|
| Figure 2e | Figure 2f |
|---|

|

|
| Figure 2g | Figure 2h |
|---|---|
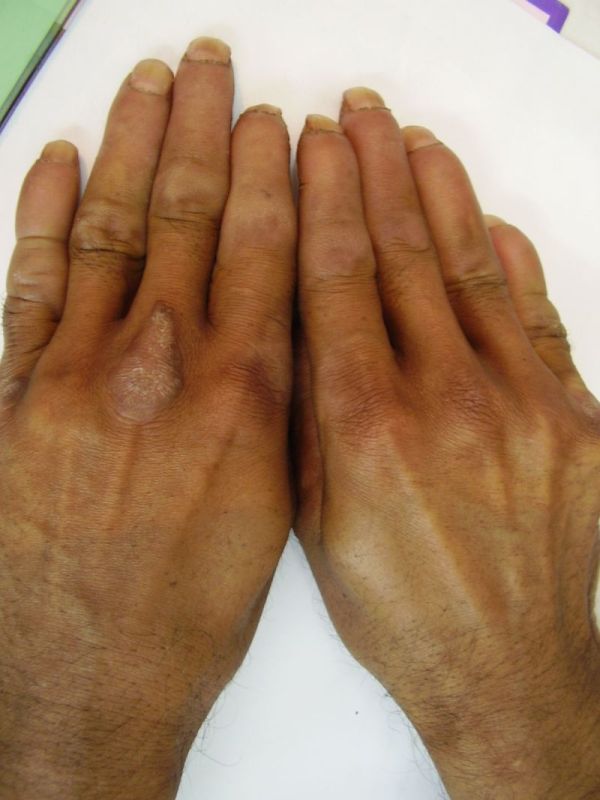
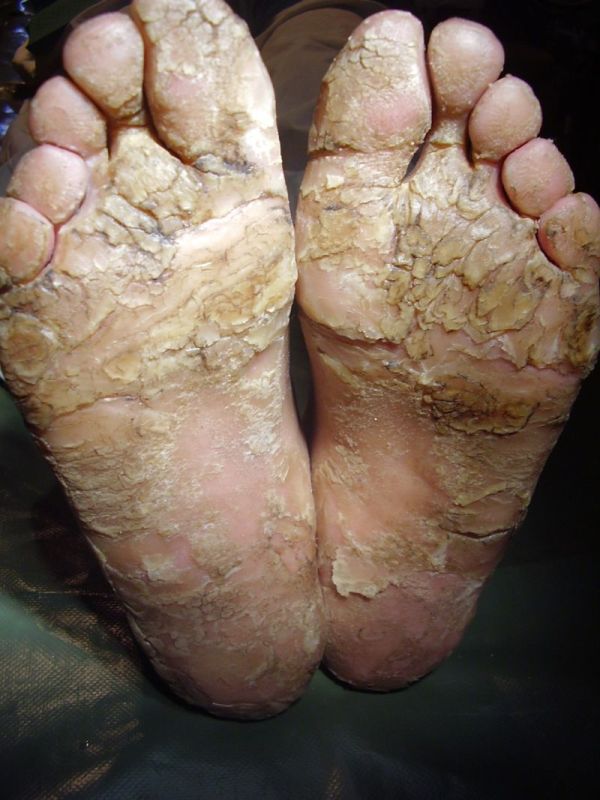
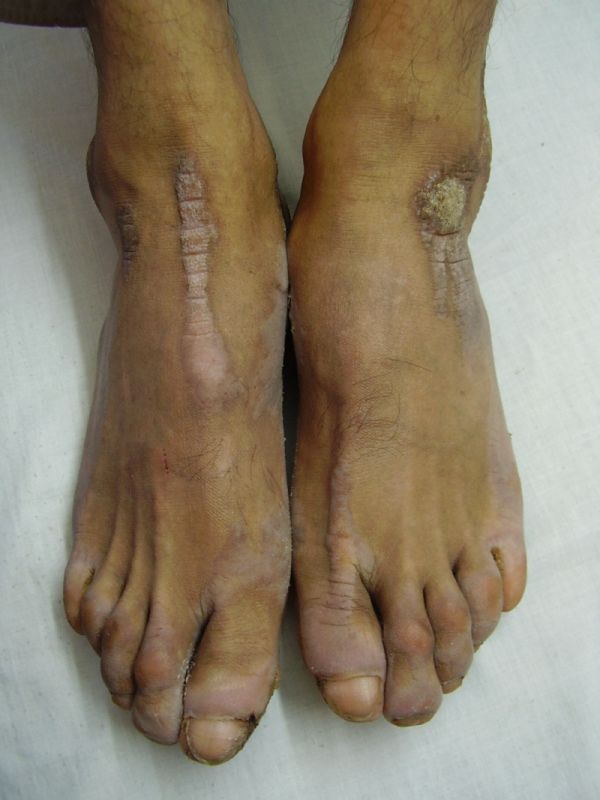
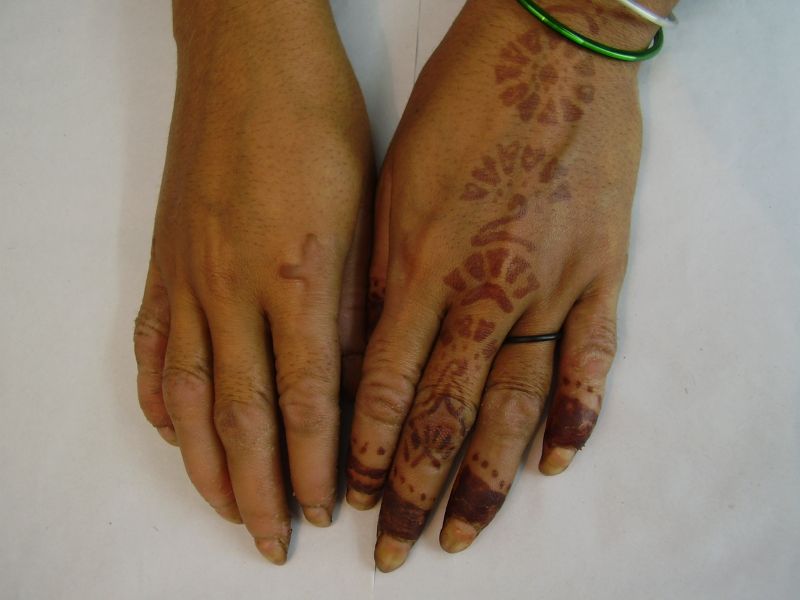
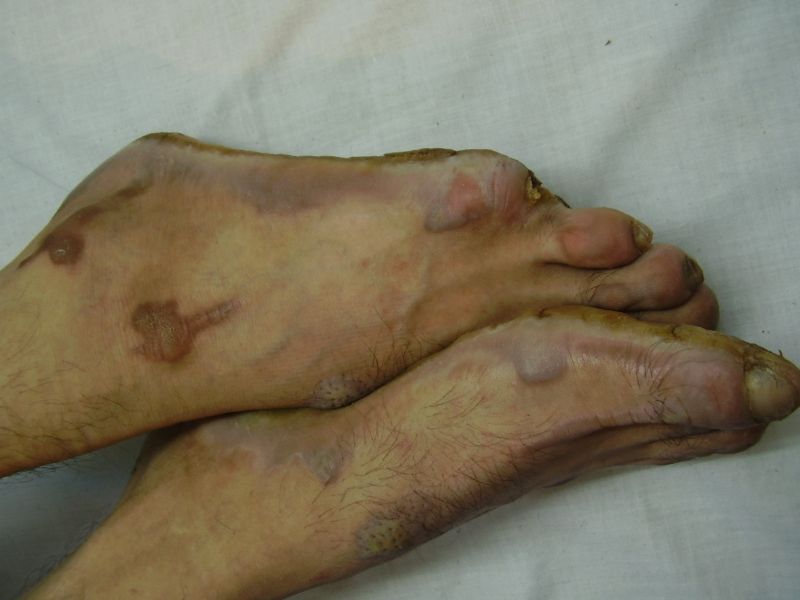
| Diffuse honeycomb palmoplantar keratoderma with marked fissures and cracks and well-defined transgredient margins (2a, 2b, 2e, 2g, 2h), hyperkeratotic starfish-like plaques over dorsal aspects of hands and feet (2c, 2f), and constriction band (pseudoainhum) around little finger (2d) in elder brother (Patient 1). | |

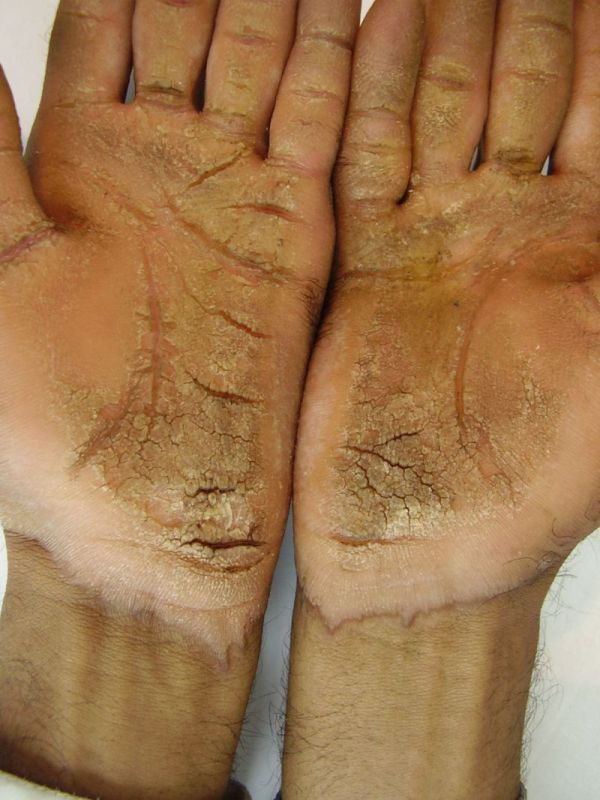
|
|
| Figure 3a | Figure 3b |
|---|
|

|
| Figure 3c | Figure 3d |
|---|

|
|
| Figure 3e | Figure 3f |
|---|---|
| Diffuse palmoplanter keratoderma with transgredient margins (3a, 3b, 3d, 3f, 3g) and hyperkeratotic plaques on dorsal hands and feet (3b, 3c, 3e) in female patient (Patient 2). | |
|
| Figure 3g |
|---|
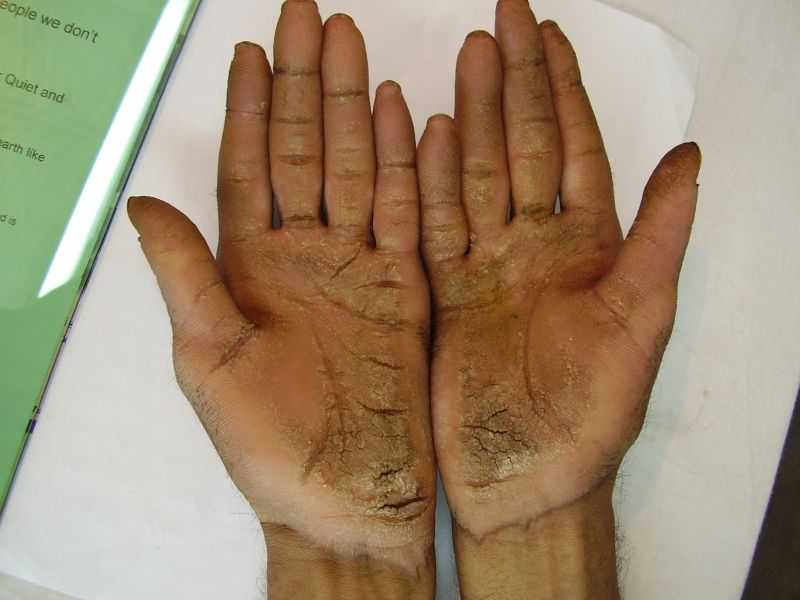
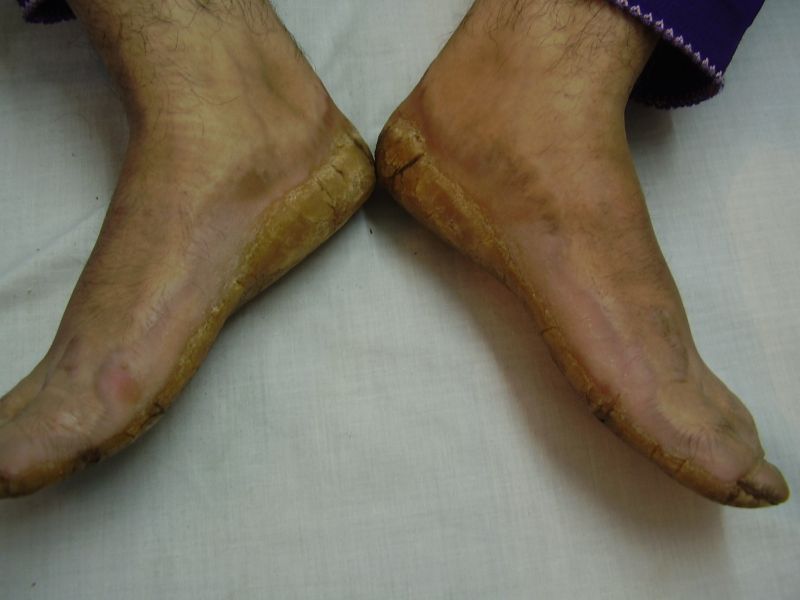
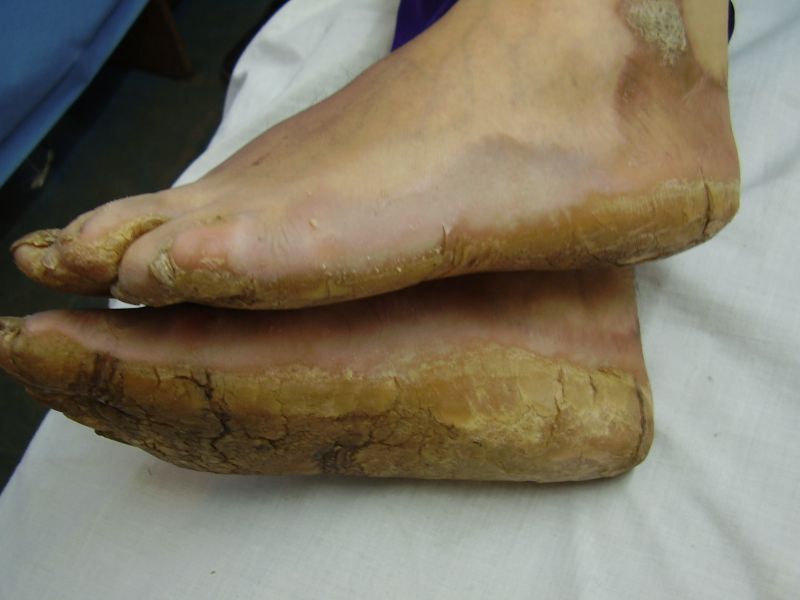
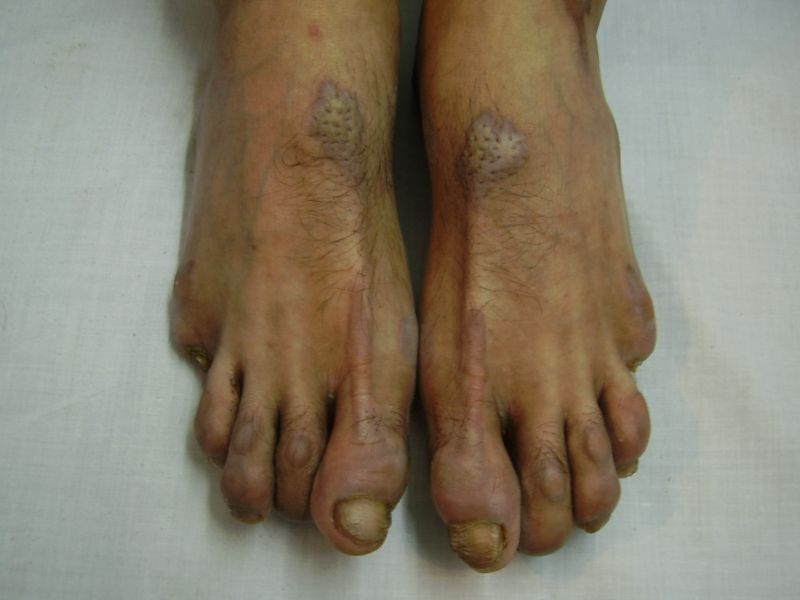
On examination, the patients showed excessive diffuse honeycomb-like thickening of the skin over the palms and soles of all hands and feet (this was more marked with deep fissures and cracks in the soles in all cases) (Figs. 1b, 2a, 2b, 2e, 2h, 3d, 4f). . The fifth toes were missing on both feet in one patient (Fig. 4f) and a constriction band was evident around little finger of left hand in another(Fig. 2d). The female patient did not yet have any signs of digital constriction bands. There was a clear-cut transgredient margin with a reddish inflammatory halo on the dorsal surfaces of palms and soles in all three patients (Figs. 1b, 2a, 2b, 2g, 2h, 3a, 3f, 3g, 4b, 4c, 4d, 4g, 4h). Starfish-like and some linear plaques were also clearly seen over the dorsal aspects of the hands and feet (Figs.1c, 2c, 2f, 3b, 3e, 4a, 4e, 4h). Hyperhidrosis of the soles was present. There was no thickening of the skin over the elbows and knees. The patients did not show any features of myopathy or spastic paraplegia. The teeth, nails, hair and mucous membranes were normal. Laboratory investigations, including complete blood count, urinalysis, and liver and renal function tests, were all within normal limits. Barium swallows did not show any abnormality. X-rays of the feet showed the absence of the fifth toes in the younger brother from autoamputation, but all other bony structures of the feet as well as the hands were without any abnormality. Audiometry was also normal. A biopsy taken from plantar skin in all patients showed massive hyperkeratosis (Fig 5) with parakeratosis, acanthosis and a sparse perivascular and periappendageal inflammatory infiltrate. There were no features suggestive of epidermolytic hyperkeratosis or any other form of dyskeratosis.
|
|
| Figure 4a | Figure 4b |
|---|
|
|
| Figure 4c | Figure 4d |
|---|
|
|
| Figure 4e | Figure 4f |
|---|
|
|
| Figure 4g | Figure 4h |
|---|---|
| Diffuse honeycomb palmoplanter keratoderma with transgredient margins (4b, 4c, 4d, 4f, 4g), hyperkeratotic plaques on dorsal hands and feet (4a, 4e) along with autoamputation of both little toes (4f) in younger brother (Patient 3). | |
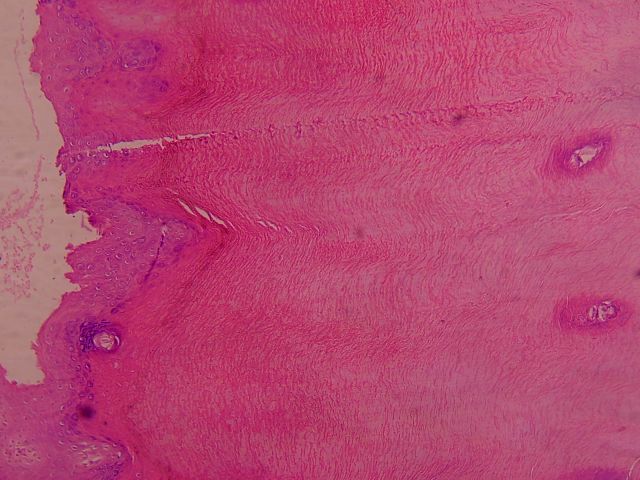
|
| Figure 5 |
|---|
| A biopsy from palm of hand. The biopsy is superficial and transects the epidermis. There is marked hyperkeratosis with focal hypergranulosis. |
The patients were reassured and provided with an explanation about the nature and course of the disease and were advised to take oral retinoids (etretinate 0.5 mg/kg body weight) along with topical keratolytics combined with emollients (10% salicylic acid in emulsifying ointment). One of the patients, who was having a painful constricting band around his left fifth finger was referred to a plastic surgeon for surgical correction of the problem.
Discussion
In 1929, Vohwinkel reported a 24-year-old woman with rapidly progressive palmo-plantar keratoderma having reddish blue border and pseudoainhum of several digits [4]. The condition was inherited in an autosomal dominant fashion. Since then there have been reports of Vohwinkel syndrome with starfish-shaped lesions over the knees and knuckles, diffuse hair loss, high frequency sensorineural hearing loss, spastic paraplegia, myopathy and ichthyosiform dermatoses. The diagnosis has largely been morphological based on the classical triad of diffuse palmoplantar keratoderma in a honeycombed pattern, constricting bands of the digits with autoamputation (pseudoainhum), and starfish-shaped hyperkeratotic plaques on the dorsum of the hands and feet, elbows, and knees (linear hyperkeratoses may also be observed on the elbows and knees) [1, 3, 5].
In our patients, there was diffuse PPK in a honeycombed pattern with transgredient extension and starfish-shaped hyperkeratotic plaques on the extensor surfaces of the hands and feet in all three patients. There was autoamputation of the fifth toes in one case and pseudoainhum was evident in another. The third patient did not yet develop any constricting bands around her digits but there was some element of sclerodactyly in all three siblings. Although none of our patients showed any other described or undescribed associations of the disease but morphologically, the features were characteristic of Vohwinkel's syndrome in all the patients. This form of progressive mutilating keratoderma has been reported to follow an autosomal dominant pattern of inheritance but in our patients none of their parents had any evidence of the disease and it was extremely unlikely to have fresh mutations in all three siblings. We think that the mode of inheritance in our cases could possibly be autosomal recessive. This recessive mode, though unusual, has occasionally been described in Vohwinkel syndrome [11]. Previously, the syndrome was mostly described in one or two siblings at the most and in one report, three generations were found to be affected [12]. To the best of our knowledge the disease has never been reported in three siblings.
References
1. Lucker GP, Van de Kerkhof PC, Steijlen PM. The hereditary palmoplantar keratoses: an updated review and classification. Br J Dermatol 1994;131(1):1-14. PubMed2. Kimyai-Asadi A, Kotcher LB, Jih MH. The molecular basis of hereditary palmoplantar keratodermas. J Am Acad Dermatol 2002;47(3):327-43. PubMed
3. Itin PH, Fistrol SK: Palmoplantar keratodermas. Clin Dermatol. 2005; 23(1): 15-22. PubMed
4. Vohwinkel KH. Keratoma hereditaria mutilans. Arch Dermatol Syphilol 1929;158:354-64.
5. Gibbs RC, Frank SB. Keratoma hereditaria mutilans (Vohwinkel). Differentiating features of conditions with constriction of digits. Arch Dermatol 1966;94(5):619-25. PubMed
6. Bergman R, Bitterman-Deutsch O, Fartasch M, Gershoni-Baruch R, Friedman-Birnbaum R. Mal de Meleda keratoderma with pseudoainhum. Br J Dermatol 1993;128(2):207-12. PubMed
7. Sharma RC, Sharma AK, Sharma NL. Pseudo-ainhum in discoid lupus erythematosus. J Dermatol 1998;25(4):275-6. PubMed
8. Maestrini E, Korge BP, Ocana-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, et al. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Mol Genet 1999;8(7):1237-43. PubMed
9. Takahashi H, Ishida-Yamamoto A, Kishi A, Ohara K, Iizuka H. Loricrin gene mutation in a Japanese patient of Vohwinkel's syndrome. J Dermatol Sci. 1999;19(1):44-7. PubMed
10. Gedicke MM, Traupe H, Fischer B, Tinschert S, Hennies HC. Towards characterization of palmoplantar keratoderma caused by gain-of-function mutation in loricrin: analysis of a family and review of the literature. Br J Dermatol. 2006;154(1):167-71. PubMed
11. Camisa C, Rossana C: Variant of keratoderma hereditaria mutilans (Vohwinkel's syndrome). Treatment with orally administered isotretinoin. Arch Dermatol 1984;120(10):1323-8. PubMed
12. Solis RR, Diven DG, Trizna Z: Vohwinkel's syndrome in three generations. J Am Acad Dermatol 2001;44(2 Suppl):376-8. PubMed
© 2006 Dermatology Online Journal