Histiocytoses
Published Web Location
https://doi.org/10.5070/D311q0g22bMain Content
Histiocytoses
VK Somani1, VNVL Sita2
Dermatology Online Journal 11 (3): 27
1. Director Somani Skin & Cosmetology Institute, Hyderabad; Formerly Professor & Head of the department of Dermatology, Deccan
College of Medical Sciences. vksomani@rediffmail.com2. Civil Assistant Surgeon, Andhra Pradesh Vaidya Vidhan Parishad, Hyderabad
Abstract
Histiocytoses are a heterogenous group of disorders that are not clearly understood. The unifying aspect of the entire spectrum lies in the accumulation of histiocytes in various tissues. Two cases are being presented for their rarity and the importance of histological features and marker studies in the diagnosis is emphasized.
Introduction
Histiocytoses are a group of poorly understood diseases characterized by accumulation of histiocytes in one or more organs leading to several disease entities often presenting with overlapping features. These are broadly classified into three categories [1],
- Langerhans cell histiocytosis (LCH)
- Non-Langerhans cell histiocytosis
- Malignant histiocytic disorders
LCH is a disease of unknown etiology and associated with a reactive proliferation of Langerhans cells in various tissues. Four main types are described, Letterer-Siwe disease , Hand-Schuller-Christian disease, eosinophilic granuloma, and Hashimoto-Pritzker disease [2]. The annual incidence of LCH has been estimated at 1 per 2,000,000 children worldwide [3].
Juvenile Xanthogranuloma (JXG) is a benign, self limiting disorder affecting mainly infants and children, characterized by accumulation of histiocytes that lack the markers of Langerhans cells. JXG is the most common form of non- Langerhans cell histiocytosis [4].
We are reporting two cases of histiocytosis, one case of Langerhans cell histiocytosis and one of non-Langerhans cell histiocytosis.
Clinical synopses
Case 1
A 2-year-old boy presented with an erythematous rash on the face, trunk, back, and scalp with crusting, oozing and scaling, of 1 year duration. Polyuria and polydypsia were present since age 8 months. The child was apparently healthy until age 1 year, although the milestones were delayed. The eruption first started on the scalp; it evolved over 1 year to involve the retroauricular area, face, trunk, and back, and was associated with mild to moderate itching. The child developed excessive thirst and micturition. There were recurrent episodes of pain and purulent discharge of both ears, swelling and bleeding gums, loss of appetite, loss of weight, and low-grade fever. No bony injuries were noted any time. The child was a full-term baby weighing 2.9 kg with no obvious congenital anomalies. Milestones were delayed with head holding at 5 months, sitting at 12 months and walking at 18 months. Nothing significant was noted regarding the family history.
On examination he was febrile, conscious, irritable, lethargic, and emaciated with abdominal distention and cervical lymphadenopathy. On cutaneous examination the scalp, retroauricular area, face, trunk and back were erythematous with multiple 2-3 mm papules with thick greasy scales and crusts. A few lesions coalesced into plaques and a few lesions were associated with oozing. Few post-inflammatory depigmented macules were present on the back. His hair was thin and lusterless. Systemic examination revealed bilateral crepitus in the lungs, hepatomegaly, swelling and bleeding gums, swelling of the hard palate, loss of incisors, and purulent discharge from the ears that was diagnosed by the otolaryngologist to be chronic otitis media.
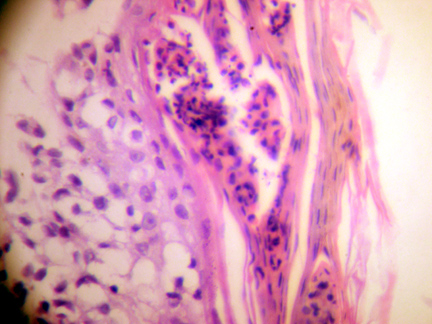
Skin biopsy showed sub-corneal pustule, spongiotic and infiltrated epidermis with vacuolated cells having large nuclei of variable sizes. A dense band-like cellular infiltrate filled the papillary and reticular dermis (Fig. 1). The cells were vacuolated with large irregular folded nuclei. Some cells had kidney shaped nuclei with eosinophilic cytoplasm (Fig. 2). Scattered eosinophils were present in the infiltrate.

|
|
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. Histopathology slide showing spongiotic epidermis, vacuolated cells with different sized large nuclei, and a dense band-like infiltrate in the entire dermis. (10 X H&E stain) | |
| Figure 2. Histopathology slide showing vacuolated histiocyte cells with large, irregular folded nuclei and some with kidney-shaped nuclei. (40 X H&E stain) | |
A tissue specimen for immunohistochemistry (Guy's King's and St Thomas' School of Medicine, St John's Institute of Dermatology, University of London) revealed diffuse positivity of tumor cells for Langerhans cell markers, S-100 and CD1a. The patient had anemia, albuminuria, reduced serum albumin, and reduced serum calcium. Urine osmolarity was always below normal (210 milli osmols/kilogram- normal 500-800 milli osmols/kilogram). Abdominal ultrasound scan showed increased density of the liver with multiple irregular lucent areas in both the lobes. The study of other organs was normal.
Skull X-ray was normal. The mastoids showed sclerosis, and both wrist joints showed osteopenia with bone age less than that of the child.
Case 2
A 1-year-old male infant was seen for brownish papulonodular eruptions on the scalp, trunk, and extremities present since the age 4 months. The lesions started as asymptomatic erythematous papules, and slowly grew over 3 months to attain the size of 1.5-2 cm. There was no itching, flushing of the face, gastrointestinal disturbances, or convulsions. Born full term to non-consanguineous parents, there were no obvious congenital anomalies. All the milestones of development were normal. Past history, family history, and birth history revealed nothing significant.
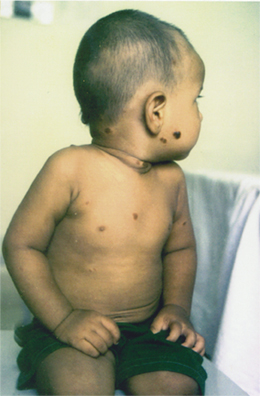
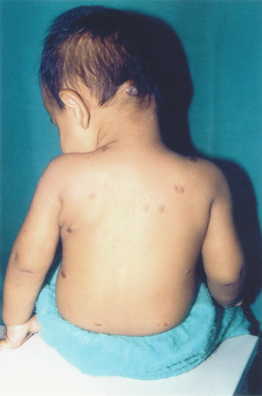
Examination revealed multiple papulonodules on the scalp, shoulders, abdomen, and back (Fig. 3). Lesions were present over the normal skin and were bluish brown, 1-2 cm in diameter, soft to firm in consistency, nonblanching to pressure, and not compressible. Few lesions showed scarring. One verrucous nodule present on the occiput bled on palpation (Fig. 4).
|
|
| Figure 3 | Figure 4 |
|---|---|
| Figure 3. Papulo nodular eruptions seen over the face, neck, trunk and occiput. | |
| Figure 4. A large nodule over the occiput along with some nodules on the back. | |
Darier sign was negative. No bony swelling or abnormality was noted. Cutaneous appendages and systemic examination were noncontributory.
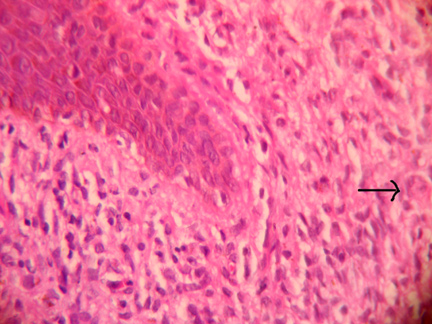
Routine investigations including chest X-Ray were within normal limits. Cutaneous biopsy showed epidermal atrophy showing no epidermotropism and a dense cellular infiltrate replacing the entire dermis extending up to the subcutaneous fat. The cells were mainly histiocytes with abundant pink, sometimes foamy cytoplasm, and vesicular nuclei admixed with poorly formed multinucleated cells, plasma cells, and lymphocytes (Fig. 5). Lipidized cells and Touton giant cells were seen in the reticular dermis (Fig. 6). A rich vascular network and extravasation of red blood cells was noted.
|

|
| Figure 5 | Figure 6 |
|---|---|
| Figure 5. Histopathology 10 X H&E stain showing dense infiltrate of histiocytes involving the entire dermis. | |
| Figure 6. Histopathology slide 40 X H & E stain showing sheets of histiocytes in the papillary dermis. Inset shows a Touton giant cell. | |
Immunohistochemistry marker studies revealed histiocyte cell marker CD68 positive, Langerhans' cell markers S 100 and CD1a, and endothelial cell marker Factor VIII a RA were negative.
Discussion
The histiocyte society describes three levels of diagnosis [1]: A presumptive diagnosis is made when the histological picture is compatible with LCH. Diagnosis is further confirmed when marker studies are done and the infiltrating cells are found to be positive for S-100 protein or peanut agglutinin. CD 1 positivity and Birbeck granules, if present, clinch the diagnosis. All the three levels were present in our first patient.
The lack of grenz zone, presence of epidermal infiltration with vacuolated cells having large nuclei of variable sizes, some cells with kidney-shaped nuclei with eosinophilic cytoplasm, and presence of eosinophils is consistent with the diagnosis of LCH. In addition to this there was S-100 and CD1a positivity leaving no room for doubt. The multisystem involvement including the mucocutaneous system, ears, oral cavity, and teeth, lungs, liver, and onset before age 2 point towards Letterer-Siwe disease. However diabetes insipidus, although one of the classic features of the triad of Hand-Schuller-Christian disease, was also present in our case.
In the second case, although lack of grenz zone and history of ulceration may suggest Langerhans-cell histiocytosis, the lack of epidermal infiltration, absence of histiocytes with reniform nuclei, and lack of eosinophils does not support this diagnosis. Further more immunohistochemical studies reveal tumor negative for S-100 and CD1a, findings that speak against histiocytosis X, congenital self-healing reticulohistiocytosis, and indeterminate cell histiocytosis. Furthermore the age of the patient, size and distribution of the lesions, and the degree of histiocytic infiltration favor multicentric xanthogranuloma. The large number of histiocytes without significant lipid accumulation favors an early lesion of juvenile xanthogranuloma [2, 5] or possibly eruptive histiocytoma. Nevertheless lipidized cells and Touton-type giant cells are present in the deeper reticular dermis favoring juvenile xanthogranuloma.
References
1. Chu A,D'Angio G,Favara B et al. Histiocytosis syndromes in children. Lancet; 208-9;1987.2. Gianotti F, Caputo R: Histiocytic syndromes: A review. J Am Acad Dermatol;13:383;1985.
3. Belaich S : Langerhans cells histiocytosis. Dermatology 1994; 189(suppl):2.
4. Cohen BA, Hood A :Xanthogranuloma: Report on clinical and histologic findings in 64 patients. Pediatr Dermatol; 6;262;1989.
5. Winkelmann RK: Cutaneous syndromes of non-X histiocytosis. A review of the macrophage-histiocyte diseases of the skin.Arch Dermatol ; 117;667;1981.
© 2005 Dermatology Online Journal