Complete form of pachydermoperiostosis: A case report

Main Content
Complete form of pachydermoperiostosis: A case report
Ram Chander MD, Sushil Kakkar MD, Arpita Jain MD, Meenu Barara MD, Kiran Agarwal MD, Bincy Varghese MD
Dermatology Online Journal 19 (2): 10
Lady Hardinge Medical College, New Delhi, IndiaAbstract
Pachydermoperiostosis is a primary form of hypertrophic osteoarthropathy, which presents with pachydermia, digital clubbing, and radiologic periostosis. Pachydermoperiostosis occurs owing to mutations of the gene encoding for 15-hydroxyprostaglandin dehydrogenase (15HPGD). Clinical manifestations of PDP are thought to relate to excessive collagen formation and dysregulation of matrix proteins because of fibroblastic hyperactivation. We present a very rare case of the complete form of pachydermoperiostosis in a young Indian male.
Introduction
Pachydermoperiostosis (PDP) is the primary form of hypertrophic osteoarthropathy (HOA) characterized by pachydermia, digital clubbing, and radiologic periostosis. It should be distinguished from the secondary form of HOA, which is much more frequent and mostly associated with severe pulmonary disease, bronchogenic carcinoma, lung empyema, bronchiectasis, congenital heart disease, and thyroid or GI malignancy [1]. This condition was first described by Friedreich in 1868, who reported this condition in two male siblings and referred to it as “hyperostosis of the entire skeleton.” In 1935, Touraine, Solente, and Gole described PDP as a primary form of HOA and distinguished its three forms. The ‘complete’ form with pachydermia and periostosis, the ‘forme fruste,’ which presents with pachydermia with minimal skeletal changes, and the ‘incomplete’ form with no pachydermia [2].
We are reporting a rare case of complete form of pachydermoperiostosis in a young Indian male.
Case report
A 20-year-old male born out of a non-consanguineous marriage, complained of deepening of skin creases over his forehead for 2 years prior to presentation. The patient noticed thickening of the skin over his forehead since early childhood, but the process had been gradual until 2 years prior at which time he noticed rapid progression and deepening of the natural skin creases over his forehead, especially over the glabellar area. Simultaneously he also noticed generalized thickening of the facial skin. The patient also had episodes of epigastric pain, which were relieved by antacids. He also complained of pain in both knee joints without any swelling or redness. He also complained of excessive sweating over his hands and feet. There were no complaints of chest pain, palpitations, dyspnea, or hemoptysis. There was no history of similar disease in the family.
 |  |
| Figure 1 | Figure 2 |
|---|---|
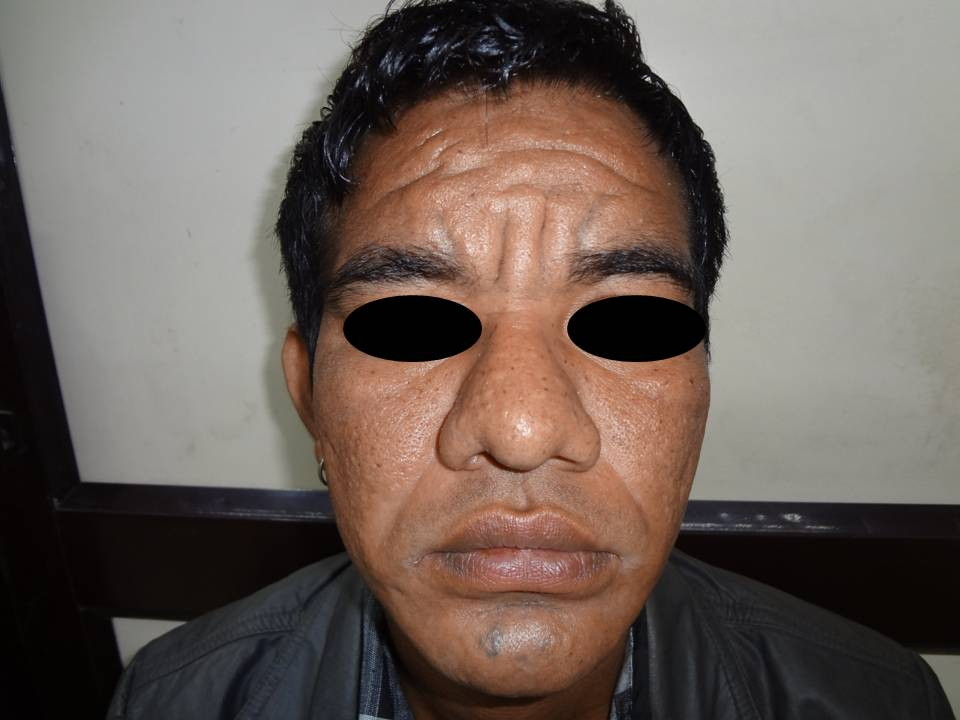
| Figure 1. Clinical photograph showing leonine facies with excessive deepening of skin creases and congested conjunctiva giving
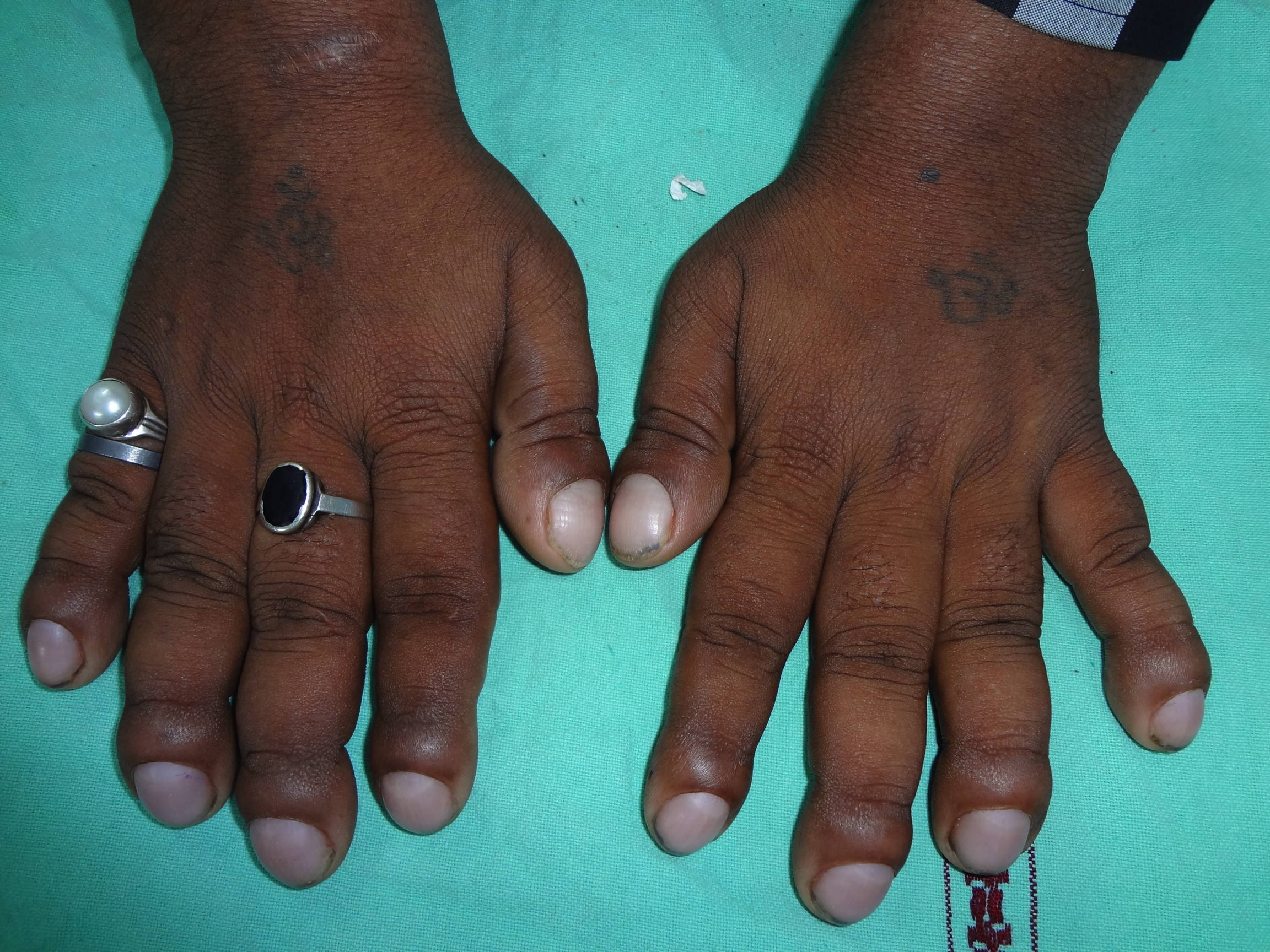
‘weary look.’ Figure 2. Clinical photograph showing short and stubby fingers with clubbing of all nails | |
On examination, the patient was comfortable with no apparent respiratory difficulty. The patient had leonine facies with excessive deepening of skin creases over the forehead, forming deep transverse and vertical grooves over the glabella (Figure 1). Both upper and lower eyelids were thickened and droopy leading to ptosis and difficulty in upward gaze. The conjunctiva was congested, which gave the patient a ‘weary looking’ facial expression. Patient also had acne along with sebaceous hyperplasia of the facial skin and coarsening of facial features. The scalp did not reveal any thickening of the skin. All his fingers were broader than normal and all the finger nails showed clubbing (Figure 2).
 |  |
| Figure 3 | Figure 4 |
|---|---|
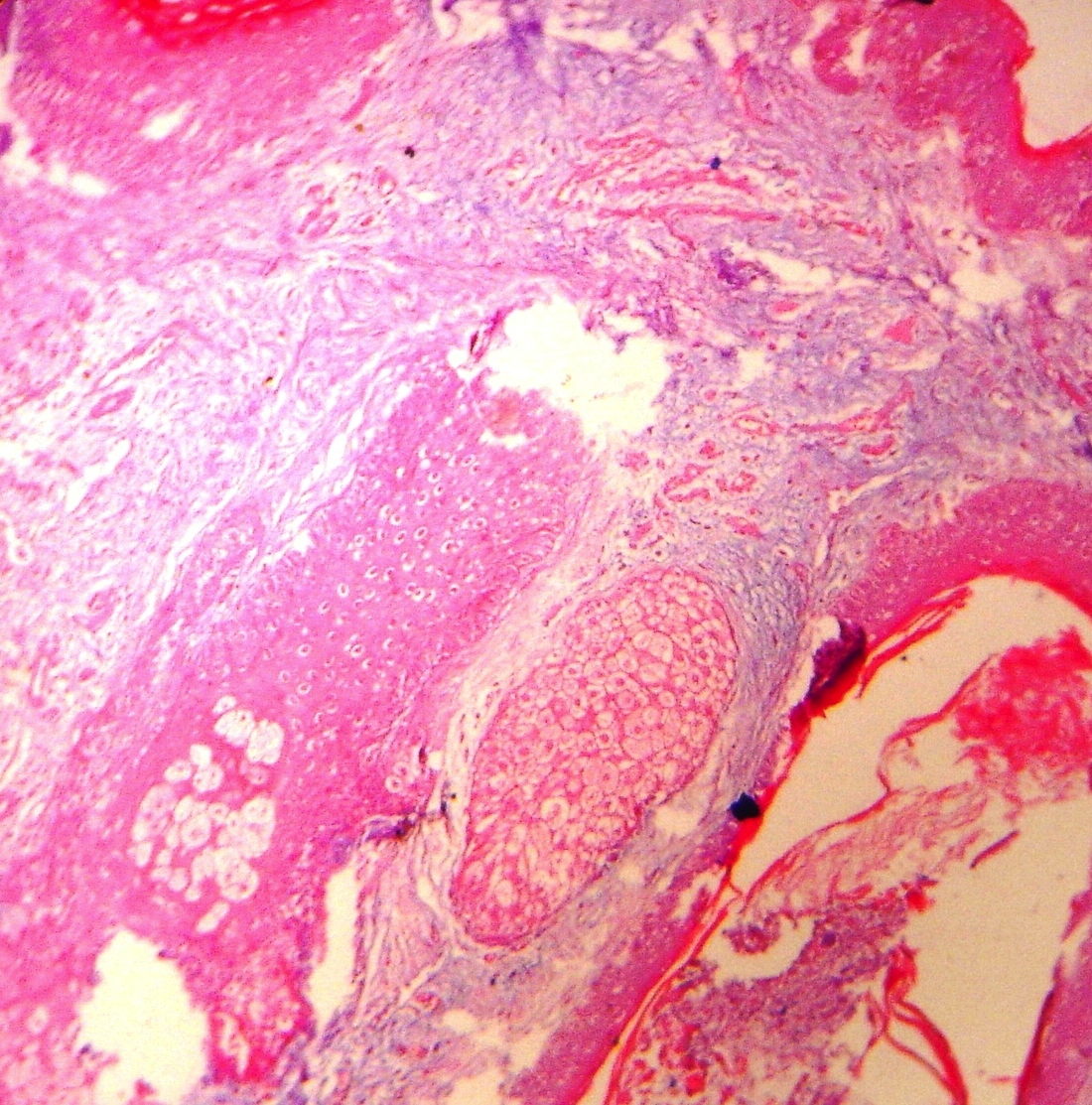
| Figure 3: Photomicrograph of face showing ground substance in the dermis, also insinuating between eccrine coils (High power
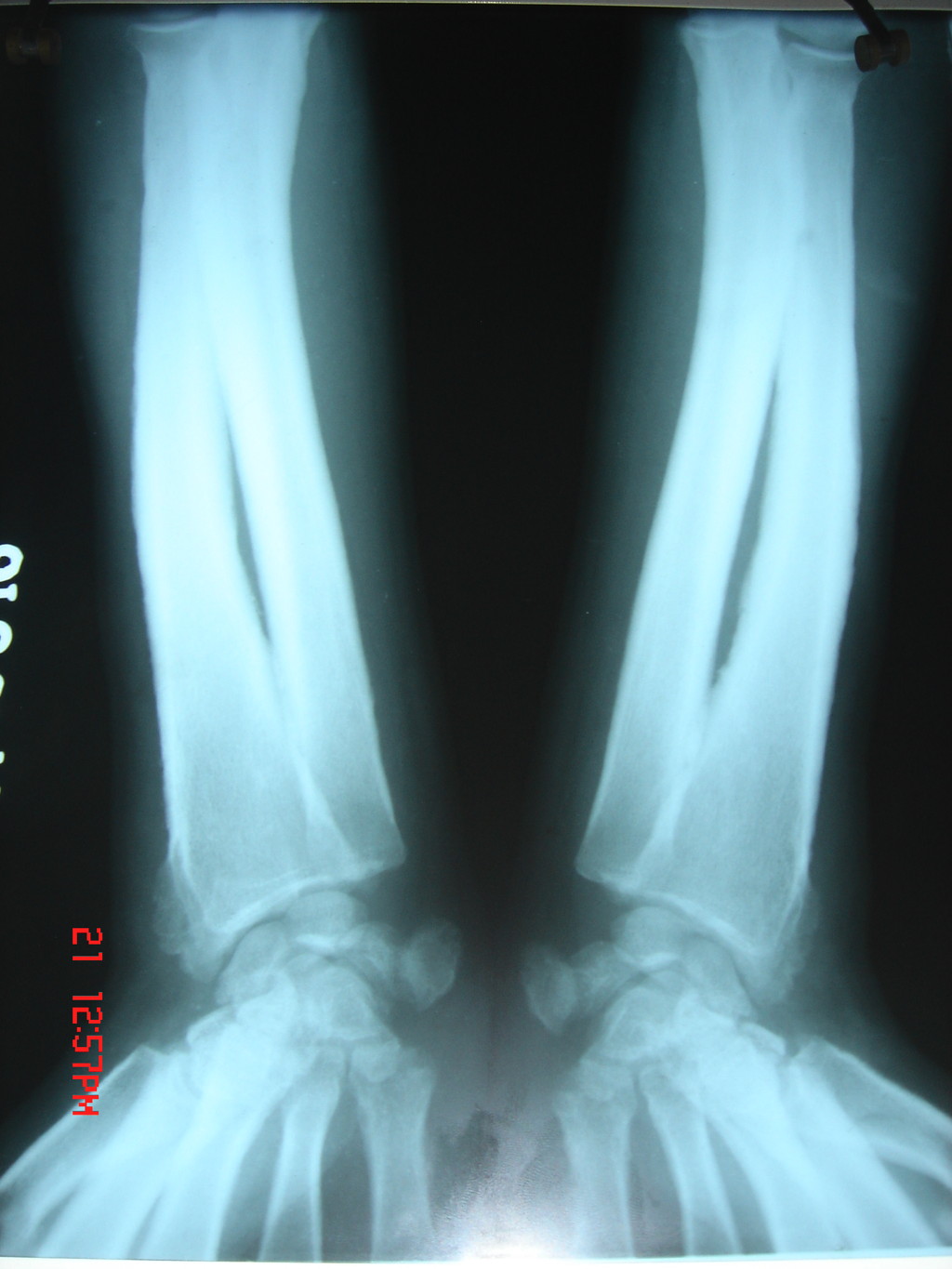
view) Figure 4. X-ray of both the legs showing periostitis of long bones | |
The diagnosis was confirmed by histopathological study, which showed an increased amount of ground substance in the dermis and insinuating between eccrine coils. It was positive for the alcian blue stain. Deep dermis showed collection of collagen bundles (Figure 3). His routine hematological investigations and thyroid function tests were within normal limits. An X-ray of hands, long bones of forearm and legs, and knee joints revealed periostosis (Figure 4). An X-ray of the hip joint showed early osteoarthritic changes. A chest X-ray showed no abnormalities in lung parenchyma but revealed thickening of clavicle and irregular lateral scapular border related to periostosis. A skull X-ray was within normal limits. Gastric endoscopy revealed a small, sessile, antral polyp over the greater curvature of the stomach. Ultrasonography of the abdomen did not reveal any abnormality. Based on pachydermia, digital clubbing, and typical radiologic findings of diffuse periostosis, a diagnosis of the complete form of pachydermoperiostosis was established.
The patient has been started on systemic retinoids (isotretinoin) in view of seborrhea, sebaceous gland hyperplasia, and coarsening of facial features. He is also taking NSAIDs for joint pain.
Discussion
PDP or Touraine-Solente-Gole syndrome is the primary form of HOA. Although an autosomal dominant inheritance with incomplete penetrance and variable expression has been confirmed, both autosomal recessive and X-linked inheritance has been suggested [3]. PDP is related to mutations of the gene encoding for 15-hydroxyprostaglandin dehydrogenase (15HPGD) [4]. PDP patients have high levels of PGE2 and decreased levels of PGE-M (the metabolite of PGE2). PGE2 can mimic the activity of osteoblasts and osteoclasts, which may be responsible for the acro-osteolysis and periosteal bone formation [5]. PGE2 also has vasodilatory effects, which may be responsible for prolonged local vasodilation resulting in digital clubbing [5]. A familial history is found in 25 to 38 percent of patients. The disorder is predominant in males and usually has its onset in late adolescence.
Clinical manifestations of PDP are thought to relate to excessive collagen formation and dysregulation of matrix proteins owing to fibroblastic hyperactivation [6]. The skin of face, forehead, and scalp become grossly thickened and are at times thrown into folds. The eyelids are thickened in 30 to 40 percent of cases giving them an expression of weariness and despair and sometimes leading to difficulty in vision [7]. Seborrhea, as seen in our case, is present in more than 90 percent with sebaceous hyperplasia, oily skin, or acne . The skin of the extremities is thickened and clubbing of the fingers is often seen. Palmo-plantar hyperhidrosis is seen in nearly 44 to 67 percent patients [7]. Sometimes patients have a spade-like appearance of the hands and feet and a cylindrical look of the arms and legs because of thickening of respective bones. Irregular periosteal ossification affects predominantly the distal ends of long bones. Associated with this is intermittent swelling or pain over large joints, which can be aggravated by alcohol intake [8]. The skin and bone changes continue to progress for 5 to 10 years, after which they usually remain unchanged throughout life. Some patients may have sparse pubic hair, gynecomastia, or periodontal defects but have normal intelligence and endocrine profile.
Upper digestive symptoms are reported by 11 to 49 percent of patients with PDP [9]. Endoscopic examination reveals gastroduodenal ulcers, atrophic gastritis, hypertrophic gastropathy, Menetrier disease, juvenile polyps, or gastric adenocarcinoma [9, 10]. Involvement of the upper digestive tract in PDP may be underestimated because most patients are asymptomatic and thus do not undergo endoscopic examination. Because PDP is often associated with gastric changes and occasionally with gastric premalignant lesions, an endoscopic follow up is indicated in patients affected with PDP who complain of upper digestive symptoms [11]. Association of PDP with juvenile polyposis has been reported in only two other patients [11, 12].
The primary form of HOA needs to be differentiated from the secondary form, which appears later in life. The secondary form is associated with some underlying pathology, presents with rapid progression, and exhibits painful skeletal symptoms. The skin findings may or may not be present in secondary cases.
Conventional PDP drug treatments to decrease inflammation and pain include NSAIDs and corticosteroids [13]. Rheumatologic symptoms can be improved by treatment with bisphosphonates, such as pamidronate or risedronate [13]. In isolated cases, tamoxifen was effective in PDP treatment, especially for bone and joint pain [13]. Retinoids are used to improve skin manifestations. Isotretinoin improves cosmetic features by inducing apoptosis within human sebaceous glands. As a result, the increase of connective tissue and hyperplasia of sebaceous glands is inhibited. Retinoids also decrease procollagen mRNA in fibroblasts, improving pachyderma [14, 15]. Colchicine can also improve skin manifestations [13]. The use of Botulinum toxin type A (BTX-A) may improve the leonine facies. Surgical methods, including facelifts and facial rhytidectomy, have also been used to improve facial appearance.
Our patient presents a complete form of pachydermoperiostosis with pachydermia, clubbing, and periosteal reaction of bones of hands, feet, and distal ends of long bones. He also had seborrhea, coarsening of facial features, hyperhidrosis, and gastric polyp. The absence of any chest complaints, a normal radiograph, and euthyroid state rule out the secondary pathologies. Normal X-ray of skull, absence of any frontal bossing, or prognathism rule out acromegaly.
The case is being reported because of the classical presentation of the rare complete primary form. Deep vertical glabellar grooving in young patients should make one investigate for this disease.
References
1. Vogel A, Goldfischer S. pachydermoperiostosis: Primary or idiopathic hypertrophic osteoarthropathy. Am J Med. 1962; 33: 166-187. [PubMed]2. Touraine A, Solente G, Gole L. Un syndrome osteo-dermopathique: la pachydermie plicaturee avec pachyperiostose des extremites. Presse Med. 1935; 43: 1820-1824.
3. Castor M, Sinibaldi L, Mingarelli R, et al. Pachydermoperiostosis: an update. Clin Genet. 2005; 68: 477-86. [PubMed]
4. Uppal S, Diggle CP, Carr IM, et al. Mutations in 15 Hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat Genet 2008; 40; 789-93. [PubMed]
5. Yuksel-KonukB, Sirmaci A, Ayten GE, et al. Homozygous mutation s in the 15 Hydroxyprostaglandin dehydrogenase gene in patients with primary hypertrophic osteoarthropathy. Rheumatol Int. 2009; 30(1): 39-43. [PubMed]
6. Martinez Lavin M, Vargas A, Rivera Vinas M. Hypertrophic osteoarthropathy: a palindrome with a pathogenic connotation. Curr Opin Rheumatol. 2008; 20: 88-91. [PubMed]
7. Kerimovic-Morina DJ, Mladenovic V. Primary hypertrophic osteoarthropathy in 32 patients. Clin Exp Rheumatol. 1992; 10: 51-56.
8. Cantatore FP, Mancini L, Ingrosso AM et al. Pachydermoperiostosis: dermatological, neurological and radiological observations. Clin Rheumatol. 1995; 14: 705-07. [PubMed]
9. Jajic I, Jajic Z. The spectrum of skeletal and visceral abnormalities in 52 patients with primary hypertrophic osteoarthropathy. Clin Exp Rheumatol. 1992; 10: 73.
10. Sethuraman G, Malhotra A, Khaitan B, et al. Familial pachydermoperiostosis in association with protein losing enteropathy. Clin Exp Dermatol. 2006; 31: 531-34. [PubMed]
11. Ikeda F, Okada H, Mizuno M et al. pachydermoperiostosis associated with juvenile polyps of the stomach and gastric adenocarcinoma. J Gastroenterol. 2004; 39: 370-74. [PubMed]
12. Mestier L de, Moreau S, Neuzillet C, et al. Gastric juvenile polyposis with high grade dysplasia in pachydermoperiostosis. Case Rep Gastroenterol. 2011; 5: 508-515. [PubMed]
13. Gomez RN, Ibanez RJ, Gonzalez PM. Primary hypertrophic osteoarthropathy. Report of 2 familial cases with literature review. Rheumatol Clin. 2009; 5: 259-63. [PubMed]
14. Athappan G, Unnikrishnan A, Chengat V, et al. Touraine Solente Gole syndrome: the disease and associated tongue fissuring. Rheumatol Int. 2009; 29: 1091-93. [PubMed]
15. Park YK, Kim HJ, Chung KY. Pachydermoperiostosis: trial with isotretinoin. Yonsei Medical Journal 1988; 29: 204-7. [PubMed]
© 2013 Dermatology Online Journal