Angiokeratoma corporis diffusum (Fabry disease)
Published Web Location
https://doi.org/10.5070/D30tt696njMain Content
Angiokeratoma corporis diffusum (Fabry disease)
Julie K Karen MD, Elizabeth K Hale MD, and Linglei Ma MD PhD
Dermatology Online Journal 11 (4): 8
Department of Dermatology, New York University School of Medicine
Abstract
A 23-year-old man presented for cosmetic consultation for symmetrically distributed, red-to-purple, hyperkeratotic papules that had been present since early childhood. Histopathologic features included ectasia of upper dermal vessels with overlying hyperkeratosis. Serum α-galactosidase A level was diminished. Fabry disease is an x-linked recessive disorder in which deficiency of the lysosomal enzyme α-galactosidase A leads to progressive accumulation of globotriaosylceramide in vital organs. The complexity and rarity of this disease mandates a multidisciplinary approach that includes initiation of enzyme replacement therapy.
A 23-year-old man, who was born of non-consanguineous parents, presented for treatment of multiple, red-purple papules that were symmetrically distributed on the lower back, hips, thighs, buttocks, and scrotum. The lesions first appeared in early childhood and had increased in number and size during the intervening years. Occasionally, an individual lesion would bleed, but they were otherwise asymptomatic. The patient, however, suffered from increasing social embarrassment because of the extensive nature of his lesions. He had previously sought dermatologic consultation, but no definitive diagnosis had been made and no treatment had been administered.
Review of systems disclosed a history of painful acroparesthesias throughout childhood; these were triggered by heat or fever, heat intolerance, and chest pain. He denied episodic fevers, abdominal pain, bone pain, arthralgias, or edema of the feet. He took no medications. The patient's sister has a few similar lesions on her fingertips, but no other family member is affected. There was no family history of mental retardation or of premature cardiovascular, cerebrovascular, or renal disease.
The patient demonstrated normal physical and mental development. Cutaneous examination showed clusters of individual, 1-5-mm, deep-red to purple papules, the largest of which were hyperkeratotic. The lesions were distributed symmetrically and involved the lower back, hips, buttocks, thighs, scrotum, and penis, with a few lesions involving the lower lip. Examination of the extremities did not show edema or varicosities. Hepatosplenomegaly was not present.
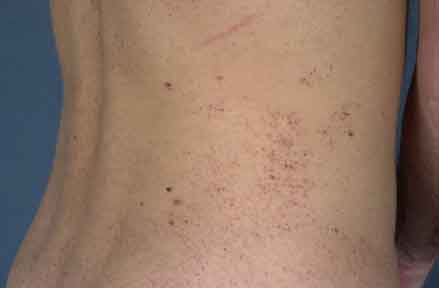
|
|
| Figure 1 | Figure 2 |
|---|
A complete blood count and comprehensive metabolic panel were normal. Serum α-galactosidase A level was 0.006U/L (reference range 0.016-0.200).
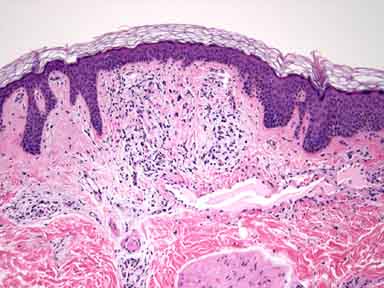
There is a proliferation of telangiectatic vessels that are lined by thin endothelial cells and a perivascular lymphocytic infiltrate. Overlying epidermis shows slight hyperplasia. Colloidal-iron stain shows deposits of connective-tissue mucin around the vessels.
Comment
The term angiokeratoma describes variably sized (punctuate-to-10-mm in diameter), hyperkeratotic papules that range in color from deep-red to blue-black. Histopathologic alterations include ectasia of upper dermal vessels with overlying epidermal hyperkeratosis.
Current classification distinguishes between localized and generalized forms. Localized angiokeratomas may be solitary or multiple, such as when distributed on the genitals (Fordyce's angiokeratoma), dorsal toes and fingers (Mibelli's angiokeratoma), or coalesced into large plaques greater than several centimeters (angiokeratoma circumscriptum naeviforme). In the generalized form, angiokeratoma corporis diffusum (ACD), lesions are usually symmetrically distributed and are mostly concentrated between the umbilicus and knees [1].
When widespread, the presence of angiokeratomas usually indicates Fabry disease, which is an inborn error of glycosphingolipid metabolism. Although rare, this x-linked recessive disorder represents the second commonest inherited lysosomal storage disease. In this disorder, deficiency of the lysosomal enzyme α-galactosidase A leads to the accumulation of globotriaosylceramide (Gal-Gal-Glc-Cer) in the vascular endothelium, smooth muscle cells, kidney, myocardium, and nervous system. The onset of symptoms is before puberty and includes acroparesthesias (periodic pain crises that involve the distal extremities), corneal and lenticular opacities, which do not impair vision, hypohidrosis or anhidrosis with associated heat intolerance, abdominal and bone pain, and cutaneous angiokeratomas. With advanced age, progressive deposition of the ceramide eventuates in early demise secondary to renal, cardiac, or cerebrovascular complications [2].
As with most x-linked recessive disorders, hemizygous men are most severely affected; however, heterozygous women have diminished enzyme levels and may develop symptoms, most commonly angiokeratomas and cataracts. A presumptive diagnosis of the hemizygous form of Fabry disease can be made by the demonstration of birefringent inclusions in the urinary sediment, corneal dystrophy on slit-lamp examination, or a skin biopsy that shows refractile lipid inclusions (so-called Maltese crosses) in endothelial cells. The gold standard of diagnosis, however, is measurement of α-galactosidase-A activity in leukocytes or fibroblasts of affected men or the detection of a mutation in the α-galactosidase-A gene in female heterozygote carriers [3].
Although previously considered a pathognomonic feature of Fabry disease, ACD is no longer regarded as specific to this disease entity. Widespread angiokeratomas also occur in patients with several additional enzyme deficiencies, which include α-fucosidase (fucosidosis), neuraminidase (sialodosis), aspartylglycosaminase (aspartylglucosaminuria), β-mannosidase (β-mannosidosis), α-N-acetylgalactosaminidase (Kansaki disease), and β-galactosidase (adult-onset GM1 gangliosidosis) [4]. The presence of one of the above conditions can be suspected on the basis of progressive psychomotor retardation, visceromegaly, facial dysmorphism, skeletal abnormalities (dysostosis multiplex), or other specific features of each disease. There have been reports of ACD occurring as a benign form without systemic features [5].
Historically, medical management was largely symptomatic—analgesics (gabapentin, carbamazepine, diphenylhydantoin) for painful episodes; renal and vascular protection with angiotensin-converting enzyme inhibitors, sodium- and protein- restricted diets, smoking cessation, and statins; and renal transplantation or hemodialysis for end-stage renal disease [6]. In recent years, enzyme replacement is a therapeutic alternative. Replacement of the deficient enzyme achieves dose-dependent clearance of the glycolipid from vital organs with consequent restoration of function [7]. The replacement enzyme, agalsidase β (Fabrazyme), received FDA approval for the treatment of Fabry disease in April 2003.
We are currently treating our patient's cutaneous lesions with the 595-nm pulsed dye (Candela V-beam) laser. Although patients with angiokeratomas may present for treatment of itching, bleeding, or superinfection (as in our patient), they seek treatment because of cosmetic concerns. Prior to the advent of laser techniques for the treatment of cutaneous vascular lesions, therapy for angiokeratomas was limited to excision, fulguration, and cryotherapy.
There are reports of successful treatment of these lesions with argon laser, copper vapor laser, variable pulse width 532-nm neodymium:yttrium-aluminium-garnet (Nd:YAG) laser, flashlamp-pumped pulsed dye laser, and potassium tritanyl phosphate (KTP) 532-nm laser [8, 9]. The Candela V-beam laser has a long pulse width that enables more uniform coagulation of the vessels, which thereby minimizes post-procedural purpura.
The complexity of Fabry disease demands a multidisciplinary therapeutic approach. We have referred our patient to Mount Sinai's General Clinical Research Center, where patients with Fabry disease undergo a comprehensive evaluation.
References
1. Grevelink SV, Mulliken JB. Vascular anomalies and tumors of skin and subcutaneous tissues. In Freedberg IM, et al., eds. Dermatology in General Medicine. 6th ed. New York: McGraw-Hill; 2003:10152. Desnick RJ, Banikazemi M. Fabry disease: alpha-galactosidase A deficiency (angiokeratoma corporis diffusum universale). In Freedberg IM, et al., eds. Dermatology in General Medicine. 6th ed. New York: McGraw-Hill; 2003:1474
3. Linthorst GE, et al. Misdiagnosis of Fabry disease: importance of biochemical confirmation of clinical or pathologic suspicion. Br J Dermatol 2004; 150: 575
4. Vargas-Diez E, et al. Angiokeratoma corporis diffusum in a Spanish patient with aspartylglucosaminuria. Br J Dermatol 2002; 147: 760
5. Marsden J, Allen R. Widespread angiokeratomas without evidence of metabolic disease (Letter). Arch Dermatol 1987; 123: 1125
6. Germain DP. Fabry disease: recent advances in enzyme replacement therapy. Expert Opin Investig Drugs 2002; 11:1467 7. Wilcox WR, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet 2004;75:65
8. Gorse SJ, et al. Successful treatment of angiokeratoma with potassium tritanyl phosphate laser (Correspondence). Br J Dermatol 2004;150:620
9. Occella C, et al. Argon laser treatment of cutaneous multiple angiokeratomas. Derm Surg 1995;21:170
© 2005 Dermatology Online Journal