Griscelli syndrome: A new phenotype with circumscribed pigment loss?
Published Web Location
https://doi.org/10.5070/D30pd3n3scMain Content
Griscelli syndrome: A new phenotype with circumscribed pigment loss?
Vidya Kharkar, Sushil Pande, Sunanda Mahajan, Ravishankar Dwiwedi, Uday Khopkar
Dermatology Online Journal 13 (2): 17
Department of Dermatology, Seth GS Medical College and KEM Hospital, MumbaiAbstract
Griscelli syndrome is a rare genetic immunodeficiency disorder characterized by pigment dilution, recurrent cutaneous and pulmonary infections, neurological deterioration, hypogammaglobulinemia, and defective cell-mediated immunity. Mutations of three distinct genes have been described in Griscelli syndrome with different phenotypes. The disease is usually fatal by the first decade of life. We report a 20-year-old female with Griscelli syndrome with circumscribed pigment loss over thighs and abdomen in addition to diffuse pigment dilution. An accelerated phase, similar to that described in Chediak-Higashi syndrome, was also observed in our case in the form of neurological deterioration. Survival of the patient beyond the first decade of life in the absence of specific therapy was also a distinctive feature.
Griscelli syndrome (GS) is a disorder of defective neutrophilic function with autosomal recessive inheritance [1]. It was first described by Griscelli in 1978, and since then only approximately 60 cases have been reported, mostly from the Turkish and Mediterranean populations [2]. The differential diagnosis of GS includes disorders of pigment dilution such as Chediak-Higashi syndrome, Elejalde syndrome, and Hermansky-Pudlak syndrome as well as disorders of neutrophilic function such as chronic granulomatous disease of childhood (CGC), myeloperoxidase deficiency, hyper-IgE syndrome and Wiskott-Aldrich syndrome. We report a 20-year-old female patient with classical features of GS along with circumscribed pigment loss (vitiligo-like lesions) and neurological involvement.
Clinical synopsis
A 20-year-old unmarried female presented with light discoloration of scalp hair, eyelashes and eyebrows since birth. The patient was born of first-degree consanguineous marriage and was the lone survivor among five siblings. All four siblings including three elderly sisters and a younger brother were born with light colored hair and expired in the first decade of life due to unexplained fever. (see Fig. 1) The patient had a history of two episodes of prolonged febrile illness due to lower respiratory tract infections in the previous year, each lasting for about 3-4 months and requiring hospitalization. She also had diplopia and headache for 4 months.
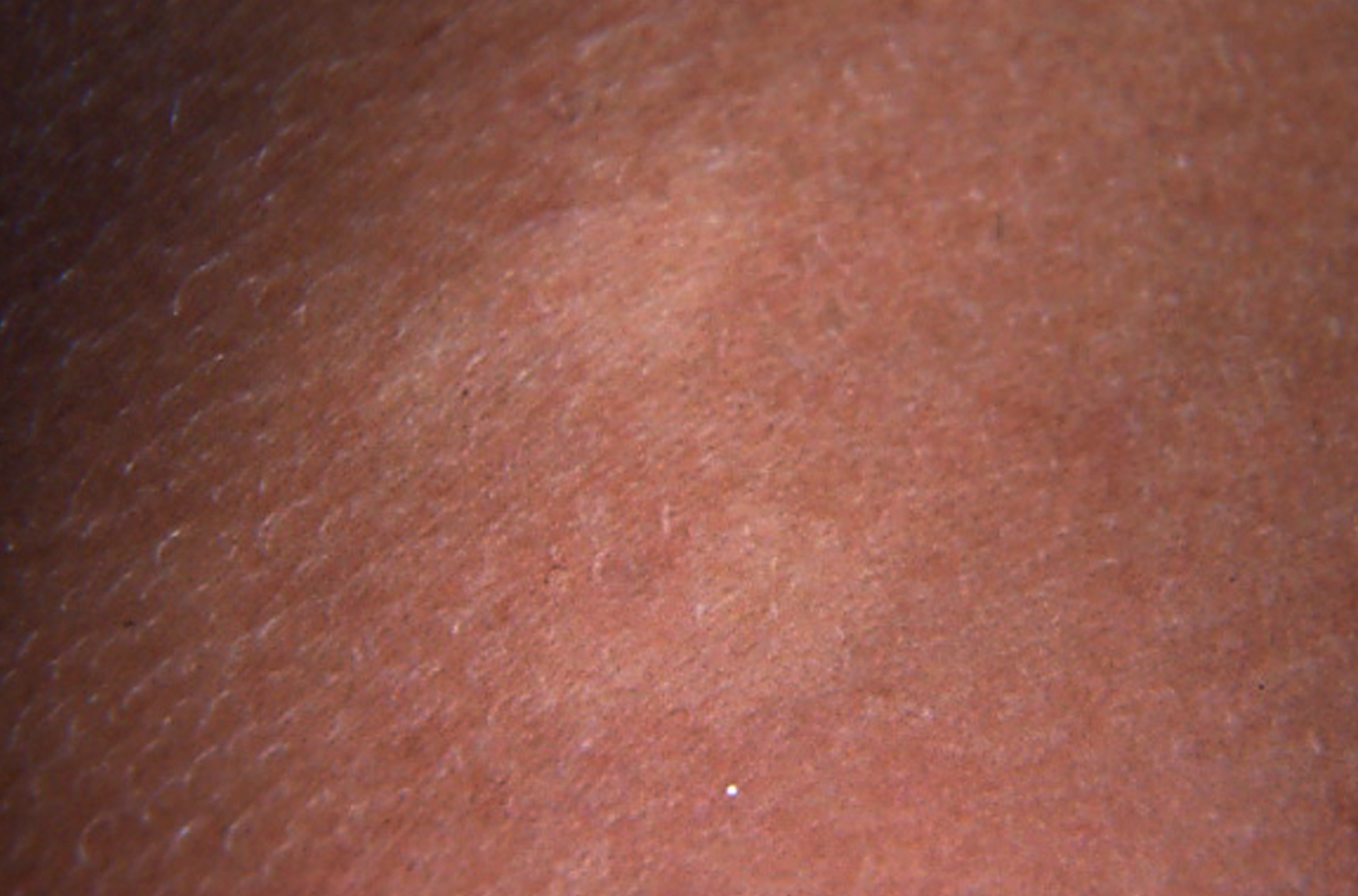 |  |
| Figure 2 | Figure 3 |
|---|---|
| Figure 2: Discrete hypopigmented macules and white discoloration of the body hair Figure 3: Large pigmentary clumps in the hair shaft | |
On examination, the scalp hair, eyebrows and body hair had a silvery shine. A few well-demarcated hypopigmented macules of 0.5 X 2 cm were scattered over the lateral thighs and abdomen (Fig. 2). There were three café-au-lait macules (approximately 1-cm diameter) on the trunk. She had diplopia on horizontal gaze in the right eye with no signs of focal neurological deficit or meningism. The remainder of her physical examination was unremarkable except for mild splenomegaly. The diagnosis of Chediak-Higashi was considered initially owing to features of pigment dilution.
Investigations revealed pancytopenia and low absolute CD4 (250/mm3 ), CD8 (126/ mm3 ), CD19 (39/ mm3 ) counts and polyclonal hypergammaglobulinemia with raised IgG (1913 mg % ), IgA (664 mg % ) IgM (416 mg % ) levels. Blood smear with Wright stain revealed normal neutrophil count without neutrophilic inclusions. Hair mount showed large pigmentary clumps in the hair shaft (Fig. 3). The patient declined a skin biopsy. Paul-Bunnel test was positive (1:8 titers). Ultrasonography of the abdomen showed mild splenomegaly. An MRI scan of brain was suggestive of white matter degeneration in pontine region and focal enhancing lesion of the suprasellar cistern of the brain. Cerebrospinal fluid (CSF) studies revealed raised total protein, few lymphocytes and absence of oligoclonal bands at 80x concentration upon CSF electrophoresis. Bone marrow studies showed normal marrow with a few plasma cells suggestive of chronic infection. Fundoscopy, skeletal survey, serum copper and ceruloplasmin levels were normal. Mitomycin stress test done to rule out Fanconi anemia was negative. Karyotyping showed a normal female 46XX pattern.
On the basis of clinical features, pigment clumping in the hair shaft, and the absence of neutrophilic inclusions on blood smears, the patient was diagnosed with Griscelli syndrome. Hemophagocytic syndrome—known to be associated with GS—was considered as the possible cause of neurological deterioration.
The patient and her relatives were counseled regarding the disease and its course. She was treated with supportive care and vitamin C 500 mg thrice daily (corrects microtubular defects in vitro) [3]. She deteriorated with the development of bilateral ocular palsies and increased headaches. Because there was no surviving sibling or HLA-identical family member, bone marrow transplantation (BMT) from HLA-identical unrelated donor was considered. However, this was not done because previous attempts of BMT from unrelated donors in GS have been unsuccessful. The patient continued to receive symptomatic care.
Comment
Griscelli syndrome is an immunodeficiency disorder characterized by partial albinism, hepatosplenomegaly, progressive neurological deterioration, hypogammaglobulinemia and pancytopenia [1]. Three different gene loci on chromosome 15q21 are responsible for this rare disease: the myosin Va gene (GS1), the RAB27A (GS2) and the MLPH (GS3) gene [4]. In the first subtype, severe neurological impairment with no immune deficiency is the characteristic feature with MyoVa mutation, as this gene regulates organelle transport in melanocytes and in neuronal cells. Immunologic abnormalities are restricted to the patients with Rab27A mutation as the capacity of the lymphocytes and NK cells of these patients to lyse target cells is impaired or absent, because of a consistent inability to secrete cytotoxic granules. Pigment dilution is common to the first two subtypes. There is evidence that a third form of GS (GS3), with expression restricted to the characteristic GS hypopigmentation, results from mutation in the gene that encodes melanophilin (Mlph) [5]. The secretory defect accounts for the hypopigmentation and the cellular immunodeficiency. Our patient had a combination of features associated with different genotypes. However, we could not confirm the mutations in our patient due to the lack of facility.
Clinical features of GS include pigment dilution (silvery white hairs, fair skin, and blue sclera). It results from aggregation of melanosomes in the melanocytes of the skin and hair shaft and their defective release to keratinocytes. Elejalde syndrome or melanolysosomal neuroectodermal syndrome is another rare autosomal recessive disorder with striking resemblance to GS that manifests with hypopigmentation, silvery hair, and early onset severe psychomotor retardation without immune deficiency [6]. Chediak-Higashi syndrome (CHS) closely resembles GS, but neurological involvement (except in accelerated phase) and large pigment clumps in hair shaft are characteristically absent in CHS. Large neutrophilic inclusions are consistently seen in CHS but not in GS. As would be expected, these were lacking in this patient. The finding of well-circumscribed hypopigmented macules in our patient remains unexplained as the patient refused skin biopsy. These could either be an incidental finding or true association with GS due to localized block in pigment transfer. Recurrent cutaneous infections were notably absent in our patient, however our patient had a history suggestive of recurrent lower respiratory tract infections. The systemic features of neurological involvement, pancytopenia, hepatomegaly, splenomegaly, and lymphadenopathy in GS are mainly due to infiltration of meninges (including brain parenchyma), bone marrow, liver, spleen, and lymph nodes by activated histiocytes- the phenomenon of hemophagocytic lymphohistiocytosis (LH) or accelerated phase. It may also be seen in Chediak Higashi syndrome. In our case, this was considered to be precipitated by Epstein Barr virus with positive Paul-Bunnel test and polyclonal hypergammaglobulinemia because the latter is not a feature of GS.
Neurological involvement—one of the important features of GS—includes white matter demyelination and lymphohistiocytic infiltration in accelerated phase as seen in our case.
In resource-limited situations, management of GS is symptomatic with counseling, ascorbic acid and non-specific immunostimulators. Systemic corticosteroids, anti-thymocyte globulins and cyclosporine are the treatment options in established cases of lymphohistiocytic histiocytosis [7]. Etoposide, corticosteroids, and intrathecal methotrexate were found to be effective with complete, but transient, remission in accelerated phase of CHS by Bejaoui et al [8].
Griscelli syndrome is a fatal disorder leading to death in early life. However genotype-phenotype correlation suggests that the natural course of the disease and outcome is dictated by the site and type of the genetic mutation. Although we could not confirm the mutation type in our patient due to lack of facilities, survival of the patient beyond the second decade in the absence of specific treatment suggests some unusual or mild form of GS. Bone marrow transplant (BMT) or peripheral blood stem cell transplantation (PBSCT) is advised as the curative therapy for GS as early as possible in the course of the disease, which suggests that the cells of hematopoietic origin are responsible for the fatal outcome in GS [9].
There are a very few reports of successful BMT in patients of Griscelli syndrome. Schuster F et al.[10] reported the first PBSCT with T cell depletion in a 6-month-old girl with deletion of the RAB27A gene. The donor was her phenotypically HLA-identical mother. Arico M et al.[11] described the first patient with GS cured with an allograft from a compatible unrelated bone marrow donor.
This is only the second case of Griscelli syndrome reported from India, but with the previously unreported features of circumscribed pigment loss amidst diffuse hypopigmentation and survival of the patient beyond the second decade of life in the absence of specific treatment.
References
1. Piette WW. Hematologic diseases. In: Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. Fitzpatrick's Dermatology in general medicine. 5th ed. New York: McGraw- Hill: 1999. p.1868.2. Sheela SR, Latha M, Susy JI. Griscelli syndrome: Rab 27a mutation. Indian Pediatrics 2004;41:944-947.
3. Bizario JC, Feldmann J, Castro FA, Menasche G, Jacob CM, Cristofani L et al. Griscelli syndrome: characterization of a new mutation and rescue of T-cytotoxic activity by retroviral transfer of RAB27A gene. J Clin Immunol. 2004;24:397-410. PubMed
4. Paller AS. Genetic immunodeficiency diseases. In: Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. Fitzpatrick's Dermatology in general medicine. 5th ed. New York: McGraw- Hill: 1999. p.1406.
5. Menasche G, Ho CH, Sanal O, Feldmann J, Tezcan I, Ersoy F, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2005;115:1100. PubMed
6. Bahadoran P, Ortonne JP, Ballotti R, de Saint Basile G. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet 2003;116A: 408-409. PubMed
7. Stephan JL, Donadieu J, Ledeist F, Blanche S, Griscelli C, Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and cyclosporin A. Blood. 1993; 15;82:2319-23. PubMed
8. Bejaoui M, Veber F, Girault D, Gaud C, Blanche S, Griscelli C, et al. The accelerated phase of Chediak-Higashi syndrome. Arch Fr Pediatr. 1989;46:733-6. PubMed
9. Schneider LC, Berman RS, Shea CR, Perez-Atayde AR, Weinstein H, Geha RS. Bone marrow transplantation (BMT) for the syndrome of pigmentary dilution and lymphohistiocytosis (Griscelli's syndrome). J Clin Immunol. 1990;10:146-53. PubMed
10. Schuster F, Stachel DK, Schmid I, Baumeister FA, Graubner UB, Weiss M, Haas RJ, Belohradsky BH. Griscelli syndrome: report of the first peripheral blood stem cell transplant and the role of mutations in the RAB27A gene as an indication for BMT. Bone Marrow Transplant. 2001 ;28:409-12. PubMed
11. Arico M, Zecca M, Santoro N, Caselli D, Maccario R, Danesino C, et al. Successful treatment of Griscelli syndrome with unrelated donor allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2002;29:995-8. PubMed
© 2007 Dermatology Online Journal