Syndromic albinism: A review of genetics and phenotypes
Published Web Location
https://doi.org/10.5070/D30fb7f671Main Content
Syndromic albinism: A review of genetics and phenotypes
Noah S Scheinfeld MD JD
Dermatology Online Journal 9 (5): 5
From the Department of Dermatology, St. Luke's-Roosevelt Hospital Center and Beth Israel Medical Center, New York. scheinfeld@earthlink.net
Abstract
There are several syndromes of albinism associated with systemic pathology. These include Chediak-Higashi Syndrome (CHS), Hermansky-Pudlack Syndrome (HPS), Griscelli Syndrome (GS), Elejalde Syndrome (ES) and Cross-McKusick-Breen Syndrome (CMBS). In the last several years the genetic defects underlying some of these syndromes have been described. HPS is related to 7 genes in humans. GS is related to 3 genes: MYOVA, Rab-27A, and melanophilin (Mlph). CHS is related to one gene: LYST. The genetic defects in ES and CMBS are yet to be defined. Syndromic forms of albinism are associated with defects in the packaging of melanin and other cellular proteins. As such they are distinct from oculocutaneous albinism, which is associated with defects in the production of melanin (e.g., TRP1, P gene, and tyrosinase).
There are several syndromes of albinism associated with systemic pathology. These include Chediak-Higashi Syndrome (CHS), Hermansky-Pudlack Syndrome (HPS), Griscelli Syndrome (GS), Elejalde Syndrome (ES), and Cross-McKusick-Breen Syndrome (CMBS). In the last several years, genetic defects underlying some of these syndromes have been described (Table 1). Moreover, variants and novel clinical phenotypes have been described as well. The common etiologic element of these syndromes seems to involve defective formation of secretory vesicles and lysosomes. This article reviews these syndromes with particular attention on newly described genetic defects and phenotypes.
Hermansky-Pudlack Syndrome (HPS)
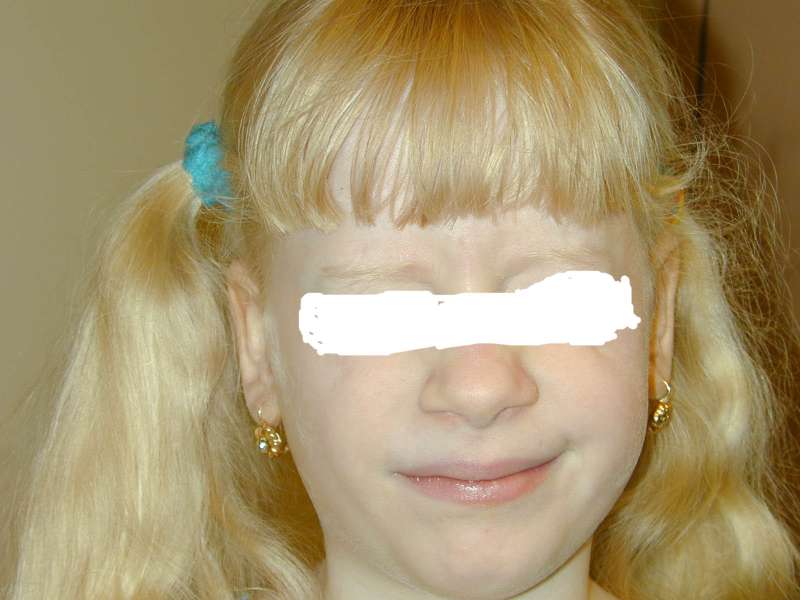
|
| Figure 1 |
|---|
| Puerto Rican girl with type-1 Hermansky Pudlack Syndrome |
HPS is very variable in presentation. It manifests with tyrosinase-positive oculocutaneous albinism, bleeding associated with platelet dysfunction, and lysosomal ceroid storage defects [1]. Visual defects occur in HPS, including photophobia, strabismus, decreased visual acuity (20/50-20/400), translucent iridies, and nystagmus. There may be epistaxis, and bruising when the child begins to walk. Pulmonary fibrosis, inflammatory bowel disease, and kidney disease occur secondary to ceroid accumulation [2].
There are now 7 distinct gene defects that are related to HPS. HPS1, HPS2 (ADTB3A), HPS3, and HPS4 are well described [3]. The genes HPS5 (ruby-eye 2) and HPS6 (ruby-eye) have recently been cloned and linked to HPS [4]. A seventh type of HPS (HPS7) has been described and is discussed below. There are probably additional gene mutations related to HPS since there are 16 mouse models of Hermansky Pudlak Syndrome [4, 5].
The HPS1 gene is located on band 10q23. A 16-base-pair frame shift duplication at exon 15 of the HPS1 is the most common defect associated with HPS in the Puerto Rican cases, and is associated with a ceroid-lipofuscin-like pigment accumulation in lysosomal structures, resulting in tissue damage [6]. A frame shift that occurs at codon 322 of HPS1 is associated with the Swiss Variant of HPS, which has a mild phenotype and normal life expectancy [7]. Other mutations exist as well. The precise role of the gene product of HPS1 remains undefined. The HPS2 gene (ADTB3A) encodes the β-3A subunit of the heterotetrameric AP3 complex [8]. HPS2 resides on chromosome 5 and assists in vesicle formation from the trans-Golgi network or late endosome. The HPS3 gene is located on band 3q24 [9]. The function of the HPS3 gene remains to be defined. HPS4 involves the human homolog of the mouse light-ear gene [10]. It is located at band 22q11.2-q12.2. HPS5 and HPS6 have been linked to orthologs of the mouse genes Ru2 and Ru (ruby-eye 2 and ruby-eye). Ru maps to chromosome 10q24.32 and RU2 maps to chromosome 11p15.4 [4]. As mentioned, a seventh type of HPS (HPS7) has been described and is discussed below.
There appear to be 3 protein complexes involved in the function and pathology of HPS. They are the HPS1/HPS4 complex BLOC-3 (biogenesis of lysosome-related organelles complex-4), adaptor complex AP-3, and BLOC-1 (biogenesis of lysosome-related organelles complex-1). These complexes are involved in the trafficking of lysosomes.
HPS1 and HPS4 proteins are part of a complex named BLOC-3 (for Biogenesis of Lysosome-related Organelles Complex 3) [5]. HPS1 and HPS4 proteins co-immunoprecipitate in vitro, indicating that they are in a complex. HPS1 and HPS4 do not interact directly in a yeast 2-hybrid system, although HPS4 interacts with itself [11]. In a partially purified vesicular/organellar fraction, HPS1 and HPS4 gene products are components of a cytosolic complex that is involved in the biogenesis of lysosomal-related organelles by a mechanism distinct from that operated by AP-3 complex [5]. Within BLOC-3, HPS1 and HPS4 are components of a discrete approximately 200-kDa module termed BLOC-4. In the cytosol, HPS1 (but not HPS4) is part of yet another complex, termed BLOC-5 [11].
The AP-3 heterotetramer complex is involved in protein trafficking to lysosomes, to melanosomes and, possibly, to platelet dense granules and Weibel-Palade bodies [12]. It has a variety of subunits that include AP3B1, which is encoded by HPS-2.
A seventh type of HPS has been defined [13]. The sdy mutant mouse expresses no dysbindin protein owing to a deletion in the gene Dtnbp1 (encoding dysbindin), and that mutation of the human ortholog DTNBP1 causes a novel form of HPS called HPS-7. Dysbindin is a ubiquitously expressed protein that binds to α- and β-dystrobrevins, components of the dystrophin-associated protein complex (DPC) in both muscle and nonmuscle cells. Dysbindin is a component of the biogenesis of lysosome-related-organelles complex 1, which regulates trafficking to lysosome-related organelles and includes the proteins pallidin, muted and cappuccino, which are associated with HPS in mice.
Genetic tests are available commercially to diagnosis HPS1 due to the 16bp duplication and the HPS3 Central PR mutation (www.medichecks.com).
Chediak-Higashi Syndrome (CHS)
Patients with CHS exhibit hypopigmentation of the skin, eyes, and hair, prolonged bleeding times, easy bruisability, recurrent infections, abnormal natural killer cell function, and peripheral neuropathy [14]. Mortality results from frequent bacterial infections or from an "accelerated phase" lymphoproliferation into the major organs of the body. By examining the DNA in mice and humans, the Beige gene on chromosome 13 in mice and the LYST gene on chromosome 1 in humans have been identified as the mutated genes in CHS. The cytoplasmic protein encoded by this gene is 3801 amino acids (425-kD). The identification of CHS1/Beige has defined a family of genes containing a common BEACH motif [15]. The BEACH motif is highly conserved in a large family of eukaryotic proteins, and is crucial for their functions in vesicle trafficking, membrane dynamics, and receptor signaling. A HEAT-repeat motif at the amino-terminus which contains several helices, and a WD40 repeat motif, which is described as a protein-protein-interaction domain, are present in the protein as well. The CHS1 gene product appears to function as an adapter protein that may juxtapose proteins that mediate intracellular membrane fusion reactions [16].
Most patients with CHS manifest with severe disease in childhood. Patients with severe childhood CHS have a functionally-null mutant CHS1 allele, whereas patients with the adolescent and adult forms of CHS have missense-mutant alleles that likely encode CHS1 polypeptides with partial function [17]. About 10-15 percent of patients exhibit a this milder clinical phenotype and survive to adulthood, but develop progressive and often fatal neurological dysfunction with intellectual decline, tremor, ataxia, peripheral neuropathy, and white-matter deterioration, often resulting in death.
Griscelli Syndrome (GS)
GS is a rare, autosomal-recessive disorder that results in pigment dilution of skin and hair (silver hair), the presence of large clumps of pigment in hair shafts, and an accumulation of melanosomes in melanocytes [18]. It is associated with defects in 3 distinct genes, RAB27A (GS2), MYO5A (GS1), and melanophilin (Mlph) (GS3), the ortholog of the gene mutated in leaden mice [19]. Children with GS associated with a defect in the RAB27A gene develop an uncontrolled T-lymphocyte and macrophage activation syndrome known as hemophagocytic syndrome (HS) or hemophagocytic lymphohistiocytosis (HLH) [20]. HS usually results in death unless the child receives a bone marrow transplant. Children with a defect in the MYO5A gene develop neurological problems but no immunological problems [21]. Slac2-a/melanophilin is the link between myosin Va (the protein product of MYO5A) and GTP-Rab27A (the protein product of RAB27A) present in the melanosome [22]. A case has been reported of GS with a defect in RAB27A that resembled Elejalde Syndrome because of the age of the patient, clinical presentation, and absence of hemophagocytosis [23].
Elejalde Syndrome (ES)
Silvery hair and central nervous system dysfunction characterize ES, a rare autosomal recessive syndrome. Elejalde Syndrome's main features include silver-leaden (silvery) hair, intense tanning after sun exposure (bronze skin color on sun-exposed areas), severe neurological impairment either congenital or developing during childhood (seizures, severe hypotonia, mental retardation) [24] and a wide spectrum of ophthalmologic abnormalities. ES does not involve impairment of the immune system. ES appears related to or allelic to GS1, and thus associated with mutations in MYOVA, however its gene mutation has yet to be defined [25].
Cross-McKusick-Breen Syndrome (CMBS)
A syndrome consisting of ocular and cutaneous hypopigmentation, severe mental retardation with spastic tetraplegia, and athetosis, was first described in 1967 by Cross in three siblings of an inbred-Amish family [26]. It has been named Cross-McKusick-Breen Syndrome (CMBS). About 10 cases of CMBS have been described, and its gene product is yet to be defined. Occurrence in siblings who are the product of parental consanguinity supports an autosomal recessive inheritance [27].
CMBS is characterized by growth retardation, dolichocephaly, cataracts, high arched palate, small, widely spaced teeth, generalized hypopigmentation, psychomotor retardation, progressive neurological manifestations, and hypochromic anemia. It has also been described with occipital cerebral atrophy, coxa valga, and generalized osteoporosis [28]. The clinical spectrum of the syndrome has been expanded to include true developmental defects of the CNS, such as cystic malformation of the posterior fossa of the Dandy-Walker type [29]. A recently reported case with classic findings also had urinary tract abnormality, bilateral inguinal hernia, focal interventricular septal hypertrophy of the heart, vacuolization of myeloid series cells, and distinct ultrastructural features of the skin [30]. Dental defects may also occur [31]. The mixed pattern of hair pigmentation is an important diagnostic sign [32]. One family has been described with two affected siblings and one sibling with silver hair who was otherwise unaffected [33]. It has been described in a Gipsy child [34] and in South Africa [35].
Other syndromic cases that manifest with albinism
There are several novel (but as yet uncategorized) syndromes with albinism. Alfadley and Parkes described 2 siblings born preterm with large ears and hypopigmented hair, who developed palmoplantar keratoderma and frontal skull bossing [37]. White reported a 20-year-old man with tyrosinase-negative oculocutaneous albinism, mental retardation, epilepsy, sensorineural deafness, ataxia, and Bartter Syndrome [37].
| Disease | Subtype | Gene | Function | Main Distinct Clinical Manifestations | Complex component |
|---|---|---|---|---|---|
| HPS | HPS1 | HPS1 | Unknown | With 16 BP deletion ceroid deposition | BLOC 3, 4 [41] |
|
|
HPS2 | ADTB3A | Secretory Vesicle formation from trans-Golgi network | Variable | AP3 |
|
|
HPS3 | HPS3 | Unknown | Usually mild symptoms |
|
|
|
HPS4 | HPS4 | Unknown | Mild symptoms to iris transillumination, variable hair/skin pigmentation, absent platelet dense bodies, occasional pulmonary fibrosis & granulomatous colitis[42] | BLOC 3, 4 [42] |
|
|
HPS5 | HPS5 | Unknown | Mild oculocutaneous albinism and easy bruising. |
|
|
|
HPS6 | HPS6 | Unknown | Oculocutaneous albinism and nosebleeds, no pulmonary or gastrointestinal symptoms, normal platelet count |
|
|
|
HPS7 | DTNBP1 | dysbindin protein in mice |
|
Biogenesis of BLOC-1 |
| CHS |
|
LYST | adapter protein for vesicle fusion | Accelerated phase lymphoproliferation |
|
| GS | GS1 | MYO5A | Myosin type protein | Neurological disease |
|
|
|
GS2 | RAB27A |
|
Hemophagocytic syndrome |
|
|
|
GS3 |
|
|
|
|
| ES |
|
Unknown |
|
Silver hair, seizures, no immune defects |
|
| CMBS |
|
Unknown |
|
Severe mental retardation with spastic tetraplegia |
|
Conclusion
Syndromic forms of albinism are associated with defects in the packaging of melanin and other cellular proteins. As such they are distinct from oculocutaneous albinism that is associated with defects in the production of melanin (e.g. TRP1, P gene, tyrosinase). The relationship of albinism and the systemic manifestations that occur in HPS, GS, CHS and ES continues to be elucidated. The link appears to be related to the cellular machinery in the ribosome involved with vesicle and lysosome production and transport. Specifically, the link between immunodeficiencies and albinism involves the use of secretory vesicles and lysosomes by the immune system [38]. Other secretory vesicles that relate to packaging of pigment are probably involved in hematological and neurological dysfunction of syndromic albinism, explaining why defects in these systems can manifest in syndromic albinism [39]. More work remains to explicate the function, identity and inter-relationship of the genes causing these diseases [40].
References
1. Krisp A, Hoffman R, Happle R, Konig A, Freyschmidt-Paul P Hermansky-Pudlak syndrome. Eur J Dermatol. 2001;11:372-3.2. Dimson O, Drolet BA, Esterly NB Hermansky-Pudlak syndrome. Pediatr Dermatol. 1999;16:475-7.
3. Ciciotte SL, Gwynn B, Moriyama K, Huizing M, Gahl WA, Bonifacino JS, Peters LL Cappuccino, a mouse model of Hermansky-Pudlak syndrome, encodes a novel protein that is part of the pallidin-muted complex (BLOC-1). Blood. 2003;101:4402-7.
4. Zhang Q, Zhao B, Li W, Oiso N, Novak EK, Rusiniak ME, Gautam R, Chintala S, O'Brien EP, Zhang Y, Roe BA, Elliott RW, Eicher EM, Liang P, Kratz C, Legius E, Spritz RA, O'Sullivan TN, Copeland NG, Jenkins NA, Swank RT. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat Genet. 2003;33:145-53.
5. Martina JA, Moriyama K, Bonifacino JS. BLOC-3, a protein complex containing the Hermansky-Pudlak syndrome gene products HPS1 and HPS4. J Biol Chem. 2003;278:29376-84.
6. Toro J, Turner M, Gahl WA. Dermatologic manifestations of Hermansky-Pudlak syndrome in patients with and without a 16-base pair duplication in the HPS1 gene. Arch Dermatol. 1999;135:774-80.
7. Schallreuter KU, Frenk E, Wolfe LS, Witkop CJ, Wood JM.Hermansky-Pudlak syndrome in a Swiss population. Dermatology. 1993;187:248-56.
8. Huizing M, Scher CD, Strovel E, Fitzpatrick DL, Hartnell LM, Anikster Y, Gahl WA.Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky-Pudlak syndrome type 2. Pediatr Res. 2002;51:150-8.
9. Huizing M, Anikster Y, Fitzpatrick DL, Jeong AB, D'Souza M, Rausche M, Toro JR, Kaiser-Kupfer MI, White JG, Gahl WA. Hermansky-Pudlak syndrome type 3 in Ashkenazi Jews and other non-Puerto Rican patients with hypopigmentation and platelet storage-pool deficiency. Am J Hum Genet. 2001;69:1022-32.
10. Suzuki T, Li W, Zhang Q, Karim A, Novak EK, Sviderskaya EV, Hill SP, Bennett DC, Levin AV, Nieuwenhuis HK, Fong CT, Castellan C, Miterski B, Swank RT, Spritz RA. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet. 2002;30:321-4.
11. Chiang PW, Oiso N, Gautam R, Suzuki T, Swank RT, Spritz RA. The Hermansky-Pudlak Syndrome 1 (HPS1) and HPS4 Proteins Are Components of Two Complexes, BLOC-3 and BLOC-4, Involved in the Biogenesis of Lysosome-related Organelles. J Biol Chem. 2003 May 30;278(22):20332-7. Epub 2003 Mar 27.
12. Starcevic M, Nazarian R, Dell'Angelica EC. The molecular machinery for the biogenesis of lysosome-related organelles: lessons from Hermansky-Pudlak syndrome. Semin Cell Dev Biol 2002;13:271-8
13. Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O'Brien EP, Tinsley CL, Blake DJ, Spritz RA, Copeland NG, Jenkins NA, Amato D, Roe BA, Starcevic M, Dell'Angelica EC, Elliott RW, Mishra V, Kingsmore SF, Paylor RE, Swank RT. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet. 2003;35:84-9.
14. Fukai K, Ishii M, Kadoya A, Chanoki M, Hamada T Chediak-Higashi syndrome: report of a case and review of the Japanese literature. J Dermatol. 1993;20:231-7.
15. Shiflett SL, Kaplan J, Ward DM.Chediak-Higashi Syndrome: a rare disorder of lysosomes and lysosome related organelles. Pigment Cell Res. 2002;15:251-7.
16. Tchernev VT, Mansfield TA, Giot L, Kumar AM, Nandabalan K, Li Y, Mishra VS, Detter JC, Rothberg JM, Wallace MR, Southwick FS, Kingsmore SF. The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Mol Med. 2002;8:56-64.
17. Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, Barbosa E, Falik-Borenstein T, Filipovich A, Ishida Y, Kivrikko S, Klein C, Kreuz F, Levin A, Miyajima H, Regueiro J, Russo C, Uyama E, Vierimaa O, Spritz RA. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet. 2002;108:16-22.
18. Mancini AJ, Chan LS, Paller AS.Partial albinism with immunodeficiency: Griscelli syndrome: report of a case and review of the literature. J Am Acad Dermatol. 1998;38:295-300.
19. Menasche G, Ho CH, Sanal O, Feldmann J, Tezcan I, Ersoy F, Houdusse A, Fischer A, de Saint Basile G. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-6.
20. Chen Y, Samaraweera P, Sun TT, Kreibich G, Orlow SJ.Rab27b association with melanosomes: dominant negative mutants disrupt melanosomal movement. J Invest Dermatol. 2002;118:933-40.
21. Sanal O, Ersoy F, Tezcan I, Metin A, Yel L, Menasche G, Gurgey A, Berkel I, de Saint Basile G.Griscelli disease: genotype-phenotype correlation in an array of clinical heterogeneity. J Clin Immunol. 2002 ;22:237-43.
22. Fukuda M, Kuroda TS, Mikoshiba K. Slac2-a/melanophilin, the missing link between Rab27 and myosin Va: implications of a tripartite protein complex for melanosome transport. J Biol Chem. 2002;277:12432-6.
23. Aksu G, Kutukculer N, Genel F, Vergin C, Omowaire B. Griscelli syndrome without hemophagocytosis in an eleven-year-old girl: Expanding the phenotypic spectrum of Rab27A mutations in humans. Am J Med Genet. 2003;116A:329-33.
24. Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, de la Luz Orozco-Covarrubias M, Tamayo L, Ruiz-Maldonando R. Elejalde syndrome--a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-6.
25. Bahadoran P, Ortonne JP, Ballotti R, De Saint-Basile G.Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116A:408-9.
26. Cross HE, McKusick VA, Breen W.A new oculocerebral syndrome with hypopigmentation. J Pediatr. 1967;70:398-406.
27. Preus M, Fraser FC, Wiglesworth FW.An oculocerebral hypopigmentation syndrome. J Genet Hum 1983;31:323-8.
28. Patton MA, Baraitser M, Heagerty AH, Eady RA. An oculocerebral hypopigmentation syndrome: a case report with clinical, histochemical, and ultrastructural findings. J Med Genet 1987;24:118-22.
29. Lerone M, Pessagno A, Taccone A, Poggi G, Romeo G, Silengo MC.Oculocerebral syndrome with hypopigmentation (Cross syndrome): report of a new case. Clin Genet. 1992;41:87-9
30. Tezcan I, Demir E, Asan E, Kale G, Muftuoglu SF, Kotiloglu E. A new case of oculocerebral hypopigmentation syndrome (Cross syndrome) with additional findings. Clin Genet. 1997;51:118-21.
31. Ozkan H, Unsal E, Kose G. Oculocerebral hypopigmentation syndrome (Cross syndrome). Turk J Pediatr. 1991;33:247-52.
32. De Jong G, Fryns JP. Oculocerebral syndrome with hypopigmentation (Cross syndrome): the mixed pattern of hair pigmentation as an important diagnostic sign. Genet Couns. 1991;2:151-5.
33. Fryns JP, Dereymaeker AM, Heremans G, Marien J, van Hauwaert J, Turner G, Hockey A, van den Berghe H. Oculocerebral syndrome with hypopigmentation (Cross syndrome). Report of two siblings born to consanguineous parents. Clin Genet. 1988;34:81-4.
34. Courtens W, Broeckx W, Ledoux M, Vamosa E. Oculocerebral hypopigmentation syndrome (Cross syndrome) in a Gipsy child. Acta Paediatr Scand. 1989;78:806-10.
35. Castle DJ, Jenkins T, Shawinsky AA. The oculocerebral syndrome in association with generalised hypopigmentation. A case report. S Afr Med J 1989 Jul 1;76(1):35-6.
36. Alfadley A, Parkes RM. Two siblings born preterm with large ears and hypopigmented hair who developed palmoplantar keratoderma and frontal skull bossing: a new syndrome? Pediatr Dermatol. 2002;19:224-8.
37. White CP, Waldron M, Jan JE, Carter JE. Oculocerebral hypopigmentation syndrome associated with Bartter syndrome. Am J Med Genet. 1993;46:592-6.
38. Griffiths GM. Albinism and immunity: what's the link? Curr Mol Med. 2002;2:479-83.
39. Huizing M, Boissy RE, Gahl WA. Hermansky-pudlak syndrome: vesicle formation from yeast to man. Pigment Cell Res. 2002;15:405-19.
40. Moriyama K, Bonifacino JS. Pallidin is a component of a multi-protein complex involved in the biogenesis of lysosome-related organelles. Traffic 2002;3:666-77.
41. Nazarian R, Falcon-Perez JM, Dell'Angelica EC. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): a complex containing the Hermansky-Pudlak syndrome (HPS) proteins HPS1 and HPS4. Proc Natl Acad Sci U S A. 2003;100:8770-5.
42. Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky-Pudlak syndrome type 4 (HPS-4): clinical and molecular characteristics. Hum Genet. 2003;113:10-7.
© 2003 Dermatology Online Journal