Posttransplant Epstein-Barr virus related lymphoproliferative disorder with a primary cutaneous presentation
Published Web Location
https://doi.org/10.5070/D30b43t7tgMain Content
Posttransplant Epstein-Barr virus related lymphoproliferative disorder with a primary cutaneous presentation
Nicholas R Snavely MD, Michael Sonabend MD, Ted Rosen MD
Dermatology Online Journal 13 (4): 7
Michael E. DeBakey Veterans Administration Medical Center, Baylor College of Medicine, Houston, Texas, USA. Abstract
Posttransplant lymphoproliferative disorder is a rare and often difficult diagnosis in patients with only cutaneous symptoms. A stepwise approach to diagnosis and classification can lead to successful treatment. We report a case of an EBV-associated posttransplant lymphoproliferative disorder (PTLD) occurring on the face with a primary cutaneous presentation. The appropriate diagnosis was made only after multiple biopsies and special stains. There was near complete resolution with decreased levels of iatrogenic immunosuppression. The diagnosis of Posttransplant lymphoproliferative disorder can be difficult to establish. A proper workup will aid in making an accelerated diagnosis and choosing appropriate treatment options.
The ever-increasing utilization of solid organ transplantation has led to the appearance of both expected and unanticipated adverse events. We report a case of one of the recently described side effects of organ transplantation (and associated iatrogenic immunosuppression), posttransplant lymphoproliferative disease. This case is particularly notable in that the disorder was confined to the skin and was therefore amenable to relatively simple therapeutic intervention consisting of decreased suppression.
Clinical synopsis
A 63-year-old man with a history of a single lung transplant in treatment of pulmonary fibrosis 5 years prior, presented to the dermatology clinic for evaluation of an acutely appearing facial lesion of approximately 1 month in duration. The completely asymptomatic lesion had been gradually increasing in size. The patient was ostensibly otherwise doing well since his transplantation. Approximately 4 years prior to presentation, the patient developed pulmonary aspergillosis, which was successfully treated with voriconazole. Current medications at the time of presentation for immunosuppression and transplant preservation included prednisone, mycophenolate mofetil, and sirolimus. For prophylaxis, the patient was taking trimethoprim-sulfamethoxazole, clarithromycin, and valganciclovir.
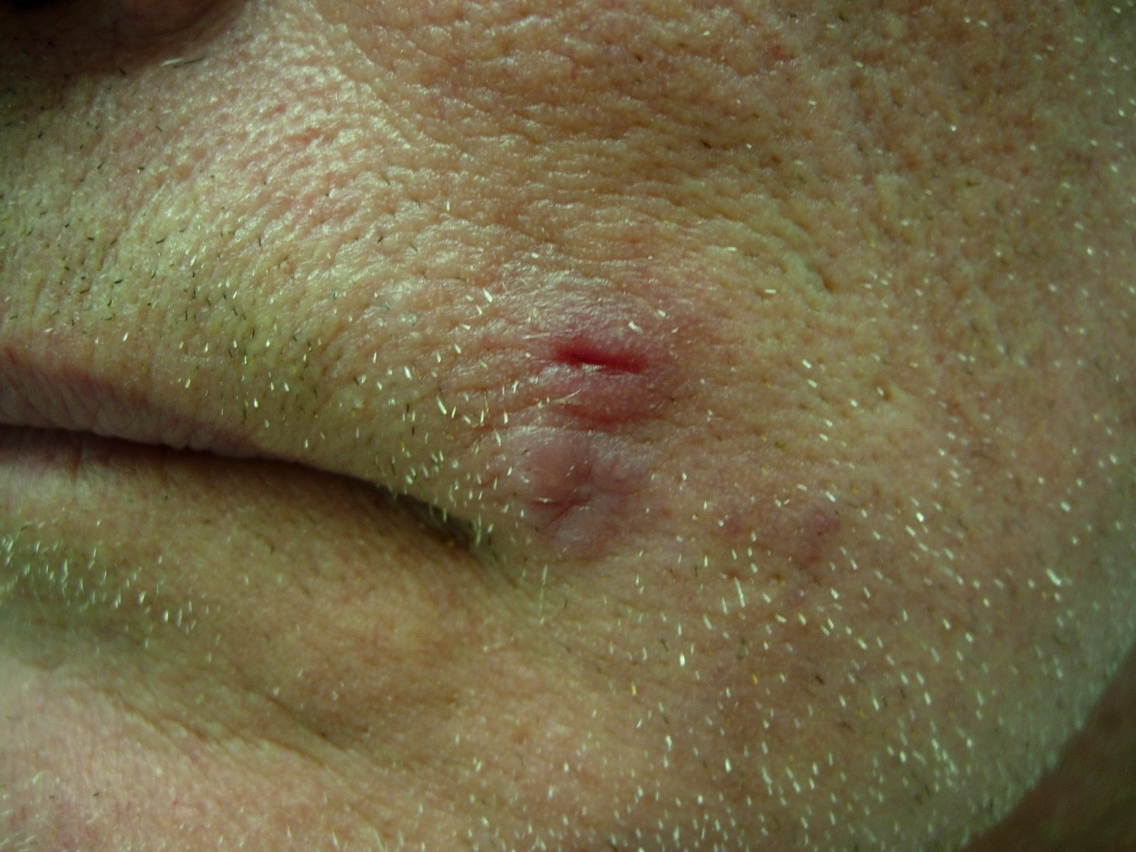
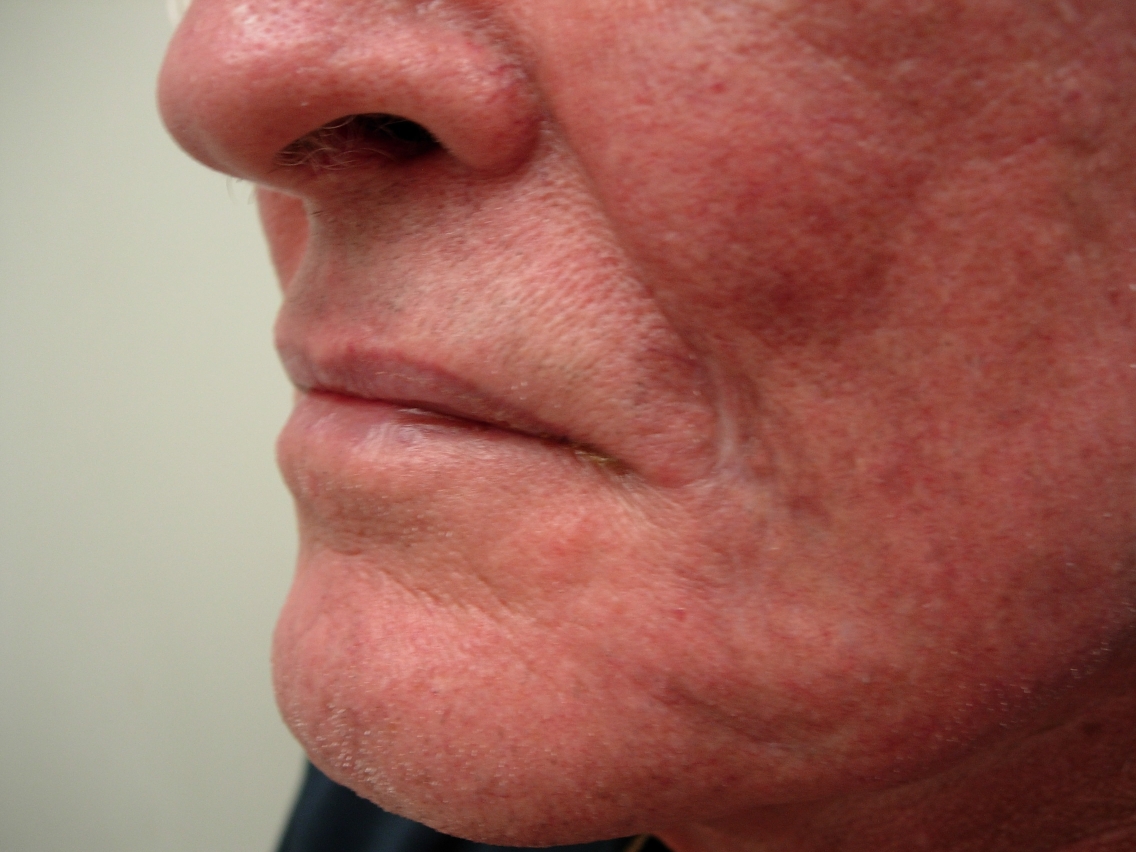
Physical examination disclosed a solitary indurated red plaque of approximately 1cm x 1cm in size located on the left cheek (Fig. 1). There were no other significant skin findings and no appreciable regional lymphadenopathy. A biopsy revealed granulomatous inflammation with a dense dermal lymphohistiocytic infiltrate, suggestive of infection. Two additional biopsies were performed for histology and culture respectively. Histology again showed well formed granulomas and a dense inflammatory infiltrate. However, special stains did not reveal any organisms and multiple tissue cultures for bacteria, fungi, and acid-fast bacilli were all negative.
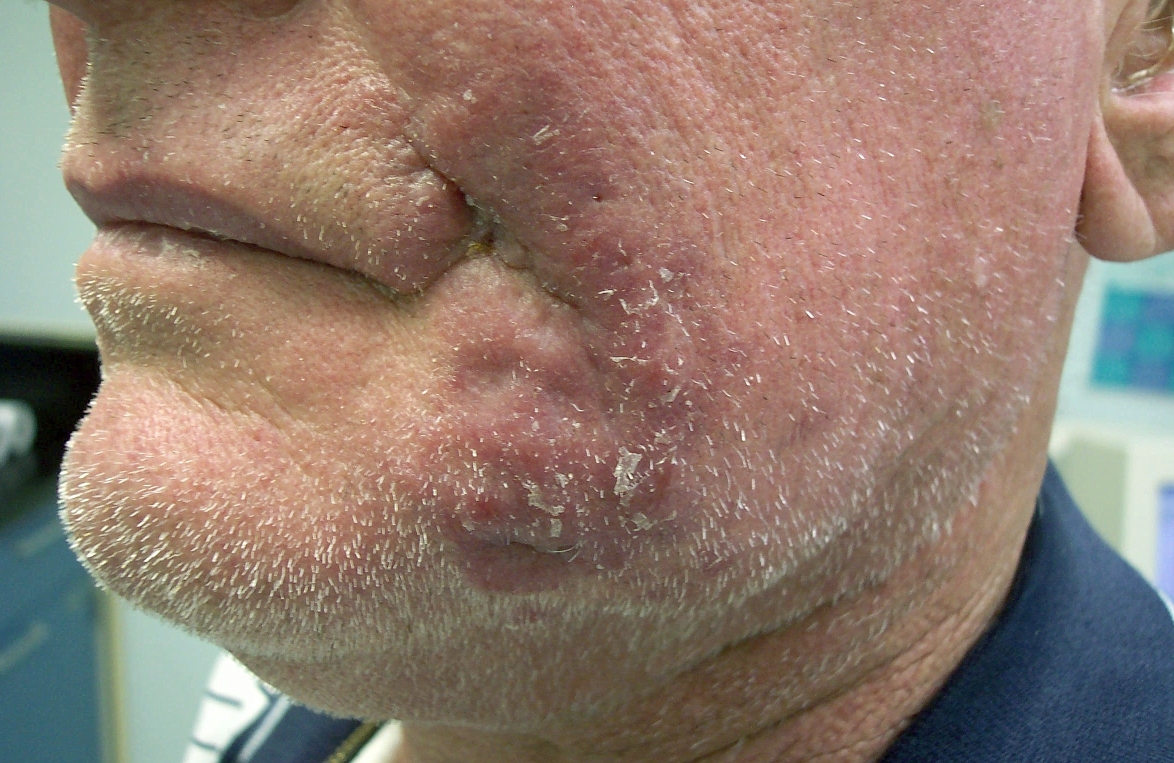
The patient's lesion continued to enlarge (Fig. 2). Four more biopsies with culture were performed, none of which were not revealing. After 6 months the patient developed a 1cm² lymph node in the left submandibular region. Needle aspiration was done on the node which showed non-caseating granulomas and no malignancy; cultures of the lymph node were negative. At the same time, a biopsy of the cheek was repeated for further histologic work-up. This biopsy again showed dense lymphohistiocytic infiltrates, extending into the subcutis. The epidermis was not involved.
 |  |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. Initial presentation of asymptomatic red plaque Figure 2. Erythematous plaque, at 4 months | |
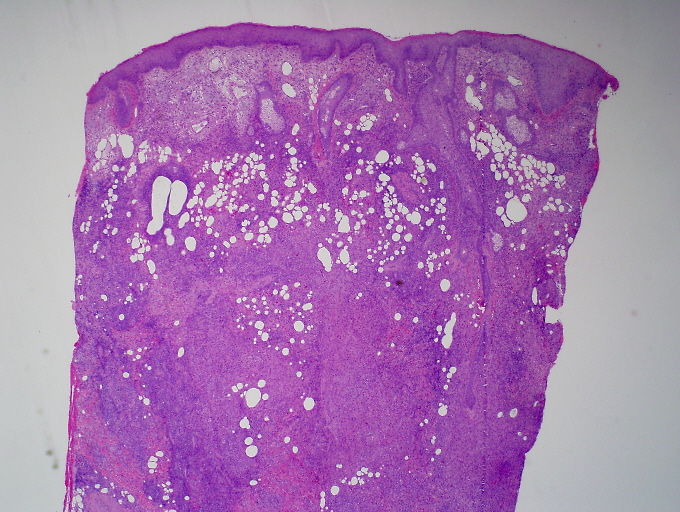
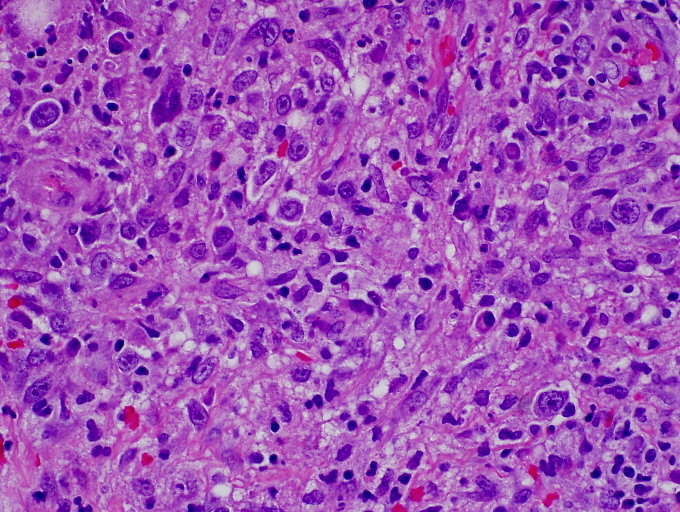
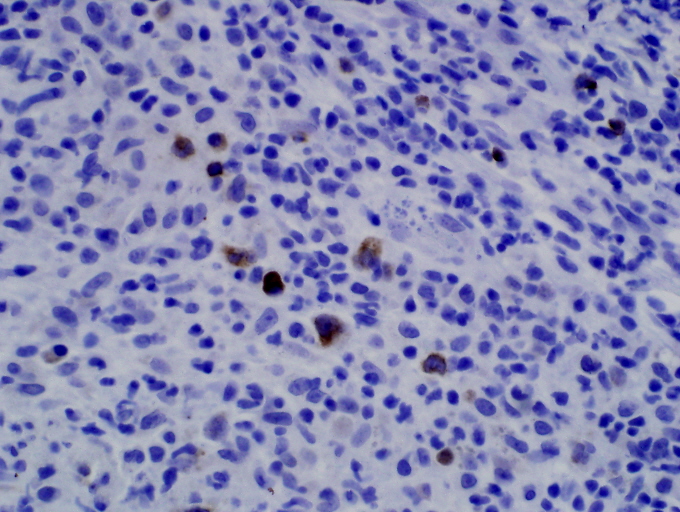
Areas of reactive germinal centers were seen, with other areas having more of a nodular or granulomatous pattern. Scattered large atypical cells were present, predominantly within collections of histiocytes. Scattered lymphocytes, plasma cells, and neutrophils were present throughout. Eosinophils were not present in significant numbers (Figs. 3 and 4). Immunohistochemical stains showed that the large atypical cells were positive for CD30, CD79a and EBV LMP, but negative for CD3, CD20, and ALK. CD45 showed diffuse positivity throughout the infiltrate (Figs. 5 and 6). No light-chain restriction was seen in the background plasma cells. Special stains for microorganisms were negative (Gram, AFB, Fite, PAS, and GMS). Diagnostic features of a lymphoma were not seen. Based on these findings, a diagnosis of EBV-related lymphoproliferative disorder was made.
 |  |
| Figure 3 | Figure 4 |
|---|---|
| Figure 3. Dense infiltrate extending throughout the dermis (H&E x 10) Figure 4. Polymorphous infiltrate including large, atypical lymphocytes, small lymphocytes, histiocytes and plasma cells (H&E x 400) | |
 |  |
| Figure 5 | Figure 6 |
|---|---|
| Figure 5: In-situ hybridization for Epstein-Barr virus (EBER) shows nuclear positivity in larger lymphocytes (400 x) Figure. 6 CD79a immunohistochemical stain demonstrating larger lymphocytes are B-cell lineage. Other large cells were predominantly macrophages (CD68 positive). | |
 |
| Figure 7 |
|---|
| Figure 7: Lesion resolved after reduced immunosuppression. |
In anticipation of a metastatic lymphoma workup, an MRI of the brain and various CAT scans were performed, all of which were normal. After consultation with the transplant team, the patient's immunosuppression was decreased: prednisone and mycophenolate mofetil were discontinued, and sirolimus dose was diminished. Remarkably, the lesion resolved within one month without further treatment (Fig. 7). At 12 months, the patient has not had any reoccurrence of the disease and his transplant remains intact.
Comments
Posttransplant lymphoproliferative disorder (PTLD) is a broad term which encompasses a range of disorders of both B and T-cell lineages, ranging from plasma-cell overgrowth to aggressive lymphomas. Such diseases can involve multiple organ systems and severity is often dependent on the immunosuppressive regimen [1]. In general, determining how aggressive a given PTLD lesion will be is based on a combination of both the clinical picture and histology, but the final behavioral outcome can be hard to predict [2]. Posttransplant lymphoproliferative disorders occur in 1-20 percent of all transplant patients, and are seen in 1.8-7.9 percent of all lung transplant patients [3]. Over 90 percent are related to EBV and are B-cell in origin. Such disorders are thought to be caused by a decreased T-cell surveillance accompanying immunosuppression, thereby allowing a reactivation of B-cells latently infected with EBV. The mainstay of treatment of PTLD has been decreasing immunosuppression; however, this maneuver has a variable and unpredictable degree of success.
Classification of PTLDs is based on a 2001 WHO summary publication that uses criteria based on systemic involvement and pathologic evaluation, especially in regards to architectural preservation and cytology [1, 3]. The WHO classification system is based on systemic categorization and is not specific to the skin [1, 4]. (Table 1) In fact, involvement of the skin in PTLD of the early and polymorphic categories is quite rare. A review of the literature reveals fewer than 20 published cases of non-lymphoma PTLD (early and polymorphic categories) with primary cutaneous presentation [4, 5, 6, 7, 8, 9].
The workup of cutaneous lymphoproliferative disorder in the posttransplant patient should include histologic and clinical correlation with an attempt to categorize the lesion according to the WHO classification system. Evaluation should begin by ruling out infectious causes of isolated lesions through multiple cultures for a broad range of organisms and via biopsy for histology accompanied by appropriate special stains. Once infection is ruled out and PTLD is favored, histologic specimens should be carefully evaluated for both cytology (monomorphic vs. polymorphic nature) and architectural disorder. Next, cell lineage should be determined. B and T cell lineage may be determined by several cellular markers. CD2 and CD3 are generally found on all T-cells, whereas B cells generally possess CD19, CD20 CD72, and CD79a [10]. CD 45 may be found on both T- and B-cell lineages [10]. CD30 has classically been expressed in anaplastic large cell lymphoma and many other anaplastic lymphoproliferative disorders, but has also been up-regulated in EBV transformed B cells [11]. Because many markers can be either up-regulated or down-regulated in the abnormal cells of PTLD, a diverse panel of antibodies is suggested for the accurate diagnosis of surface markers in order to attempt delineation of cell lineage [2].
Clonality testing is useful in the work-up of PTLD. Clonality of B-cell or T-cell lineages is best determined by the finding of clonal immunoglobulin gene re-arrangements by PCR or by detection of clonal EBV in the tumor by PCR. Detection of light chain restriction by immunohistochemistry is an easier, although less reliable indicator of clonality [12]. In a study by Wood et al. 14 cases of pseudo-B-cell lymphoma in which all, by definition, had polyclonal immunoglobulin light chains (no restriction), five were actually shown to be monoclonal by PCR [12]. Therefore, even though lack of light chain restriction by immunohistochemistry may lend weight to the polyclonal nature of an infiltrate, the gold standard is immunoglobulin gene rearrangement studies by PCR on fresh-frozen tissue.
Presence of EBV should be evaluated in all cases of suspected PTLD. Detection should be based on in-situ hybridization of 1) the non-translated RNA latent oncogene produced by EBV called EBER, 2) the EBV Latent Membrane Protein 1 (LMP-1), or by 3) EBV PCR (which can also help demonstrate clonality of the cellular infiltrate) [13]. Diagnosis of EBV using serology (i.e., heterophile Ab/monospot test or viral specific Ag serology) is not an accurate indicator of infection in posttransplant patients due to the anergy caused by immunosuppression [14]. It is worth noting that the quantitative measurement of Epstein-Barr viral load in the whole blood of patients with PTLD provides no predictive information as to ultimate clinical course [15].
Some prognostic information can be gained by looking for certain mutations in affected cells. Some of the more aggressive forms of PTLD have shown mutations in RAS, and MYC oncogenes, but these de-differentiations are rare in less aggressive polymorphic varieties of disease [3]. BCL6 mutations have been documented in 40-90 percent of polymorphic and monomorphic posttransplant lymphoproliferative disorders, and the BCL6 mutation has been shown to correlate with a decreased response to reduced immunosuppression [3]. Therefore, BCL6 should be evaluated in even the most benign appearing forms of PTLD.
Our patient was given the diagnosis of primary cutaneous posttransplant lymphoproliferative disorder. We further classified his disease as polymorphic variant because of the location of the lesion, total lack of systemic symptoms, and histologic features. Although fresh-frozen tissue for PCR analysis was not available to confirm it, immunoglobulin tests suggest a lack of B-cell monoclonality further classifying our lesion as polymorphic, polyclonal PTLD. Epstein-Barr virus was found by EBER in-situ hybridization. B-cell lineage was confirmed due to the CD3 negativity and CD79a positivity of immunohistochemical stains. Our patient was appropriately treated with decreased immunosuppression with subsequent resolution of the lesion. Because our patient responded to decreased immunosuppression, no further treatment was necessary, but other treatment methods such as interferon-&alfa;, monoclonal-antibody therapy with rituximab, EBV-specific cytotoxic T-cell infusion, multi-agent systemic chemotherapy and preventative vaccines with donors have all been suggested [13]. Treatment with acyclovir, which inhibits replication of EBV in latently infected cells, is generally not effective for PTLD [13]. Despite any and all of the aforementioned maneuvers, some cases of PTLD do not respond, and the overall five-year survival rate remains relatively low [16,17].
By utilizing the sequential, stepwise approach enumerated above, establishing a diagnosis and further categorizing PTLD is possible. Timely diagnosis allows for rapid and proper intervention. Ultimately, some lesions elude strict delineation at the time of presentation, and disease may best be characterized in retrospect by the final biologic behavior.
References
1. Harris NL, Swerdlow SH, Frizzera G, Knowles DM. Post-transplant lymphoproliferative disorders. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, eds. Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France; IARC Press; 2001. Pages 264-269.2. Ploysangam T, Breneman D, Mutasim DF. Cutaneous pseudolymphomas. J Am Acad Dermatol 1998;38; 877-92.
3. Andreone P., Gramenzi A, Lorenzini S, Biselli M, Cursaro C, Pileri S, Bernardi M. Posttransplantation lymphoproliferative disorders. Arch Intern Med 2003; 163:1997-2004
4. Capaldi L, Robinson-Bostom L, Kerr P, Gohh R. Localized cutaneous posttransplant Epstein-Barr virus-associated lymphoproliferative disorder. J Am Acad Dermatol 2004;51:778-80.
5. Gonthier DM, Hartman G, Holley JL. Posttransplant lymphoproliferative disorder presenting as an isolated skin lesion. Am J Kidney Dis 1992;19; 600-03.
6. Chai C, White WL, Shea CR, Prieto VG. Epstein Barr virus-associated lymphoproliferative-disorders primarily involving the skin. J Cutan Path 1999: 26:242-47.
7. Schumann KW, Oriba HA, Bergfeld WF, Hsi ED, Hollandsworth K. Cutaneous presentation of posttransplant lymphoproliferative disorder. J Am Acad Dermatol 2000; 42; 923-26.
8. Ahmed I, McEvoy MT. Cutaneous B-cell post transplant lymphoproliferative disorder: report of 7 cases. J Cutan Pathol 2000; 27:547. (Abstract only)
9. Blokx W, Andriesesen M, van Hammersvelt HW, van Krieken JH. Initial spontaneous remission of posttransplantation Epstein Barr virus-related B-cell lymphoproliferative disorder of the skin in a renal transplant recipient. Case report and review of the literature on cutaneous B-cell posttransplantation lymphoproliferative disease. Am J Dermatopathol 2002;24; 414-22.
10. Weeden D. Skin Pathology. London; Churchill Livingstone, 1997. Pages 216-230 and 897-921.
11. Kahn A, Horenstein M. Inflammatory Dermatopathology. The diagnosis of cutaneous inflammatory infiltrates with atypical CD30-positive cells. Pathol Case Rev 2004;9; 60-5.
12. Rijlaarsdam JU, Willemze R. Cutaneous pseudolymphomas: classification and differential diagnosis. Semin Dermatol 1994;13;187-96.
13. Cohen, JI. Epstein-Barr virus infection. N Engl J Med 2000;343; 481-492.
14. Ikediobi NI, Tyring SK. Cutaneous manifestations of Epstein-Barr virus infection. Dermatol Clin. 2002;20:283-9
15. Oertel S, Trappe RU, Zeidler K, Babel N, Reinke P, Hummel M, Jonas S, Papp-Vary M, Subklewe M, Dorken B, Riess H, Gartner B. Epstein-Barr viral load in whole blood of adults with posttransplant lymphoproliferative disorder after solid organ transplantation does not correlate with clinical course. Ann Hematol 2006; 85; 478-84.
16. Jain AB, Marcos A, Pokharna R, Shapiro R, Fontes PA, Marsh W, Mohanka R, Fung JJ. Rituximab (chimeric anti-CD20 antibody) for posttransplant lymphoproliferative disorder after solid organ transplantation in adults: long-term experience from a single center. Transplantation 2005;80; 1692-98.
17. Lorenzini S, Andreone P, Gramenzi A, Morelli C, Zinzani PL, Grazi GL, Pileri S, Baccarani M, Tura S, Bernardi M. Posttransplant lymphoproliferative disorders in liver transplanted patients: a report of four cases. Transplant Proc 2006;38; 1477-80.
© 2007 Dermatology Online Journal