Commentary on Degos Disease A C5b-9/Interferon-α-Mediated Endotheliopathy Syndrome by Magro et al: A reconsideration of Degos Disease as hematologic or endothelial genetic disease
Published Web Location
https://doi.org/10.5070/D309r396d1Main Content
Commentary on Degos Disease A C5b-9/Interferon-α-Mediated Endotheliopathy Syndrome by Magro et al: A reconsideration of Degos
Disease as hematologic or endothelial genetic disease
Noah Scheinfeld MD
Dermatology Online Journal 17 (8): 6
Assistant Clinical Professor of Dermatology, Columbia University, New York City, New YorkAbstract
Magro et al in April of 2011 published a new article in the American Journal of Clinical Pathology on the etiology and treatment of Degos Disease (DD), and importantly, its fatal variant malignant atrophic papulosis (MAP). Specifically, Magro noted that MAP is a disease involving the complement cascade that can be treated effectively with eculizumab. DD has two variants, a benign variant confined to the skin and a malignant (heretofore fatal) variant that involves the skin and systemic organs. Five aspects of DD are discussed: (1) the clinical findings of DD, (2) thrombosis and DD, (3) the histology of DD, (4) the presence of viral like inclusions in the endothelial cells of patients with DD, and (5) the lack of any apparent immune defect that relates to DD. It seems the previous criteria for Degos Disease must be amended. Paroxysmal nocturnal hemoglobinuria (PNH) is discussed and its relationship with DD explored. Eculizumab has been approved to treat paroxysmal nocturnal hemoglobinuria. A review of the data suggests that MAP is a hematological or endothelial disease like PNH. PNH, eculizumab, and data about DD is discussed to give a basis for understanding DD and speculate why eculizumab may be promising for the treatment of MAP.
Introduction
With the publication of Magro et al (2011) [1] concerning the successful treatment of Systemic Degos Disease (SDD) (also known as Malignant Atrophic Papulosis or MAP) with eculizumab, physicians have entered into a new age of understanding this condition. Further, with the explication of the role of complement in the pathogenesis and perpetuation of MAP researchers have crossed the threshold into a new milieu for defining MAP.
It must be recalled as we discuss MAP that the disease that Degos (1942) [2, 3] and Kohlmeier (1941) [4] described has two broad variants, one benign and the other malignant. The benign variant is confined to the skin and is referred to as benign atropic papulosis (BAP), localized Degos Disease (LDD), or cutaneous Degos Disease (CDD). The malignant (formerly fatal) variant [7] involves the skin and systemic organs (most commonly the gastrointestinal tract and central nervous system) and is referred to as malignant atrophic papulosis (MAP) or systemic Degos Disease (SDD). Together, these two entities are most commonly called Degos Disease (DD). Most cases of eruptions with the clinical and histological findings of DD are benign atrophic papulosis and are confined to the skin alone [5, 8]. The complete mechanisms that underlie Degos Disease broadly, the limitation of BAP to the skin, the benign course of BAP, and the fatal course of MAP have yet to be defined even now in June of 2011 after the publication of Margo's article in April of 2011.
Five Important Aspects of Degos Disease
To open my discussion of DD, I will discuss 5 subjects: (1) the clinical findings of DD, (2) thrombosis and DD, (3) the histology of DD, (4) the presence of viral like inclusions in the endothelial cells of patients with DD, and (5) the lack of any apparent immune defect that relates to DD [6].
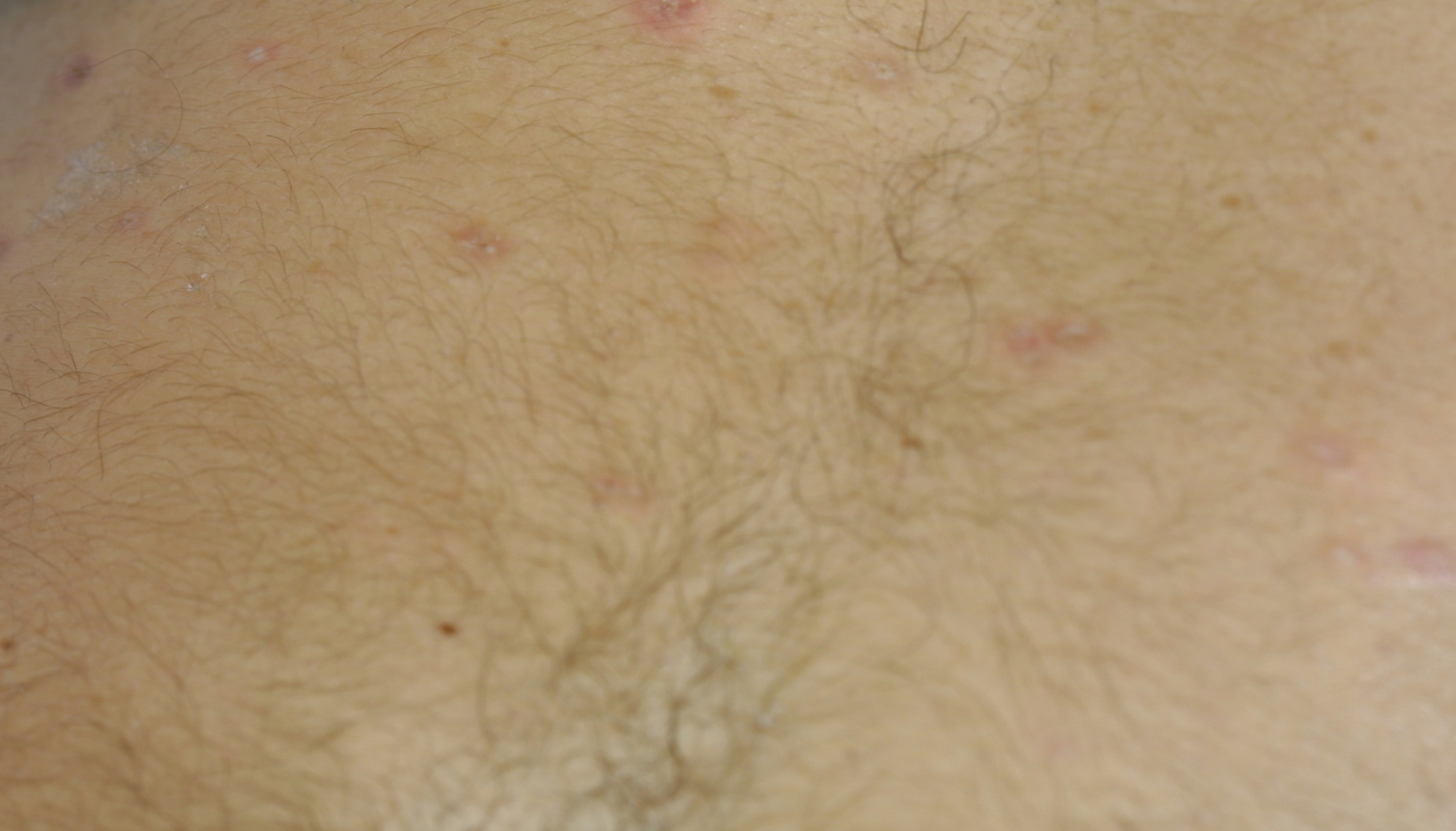 | 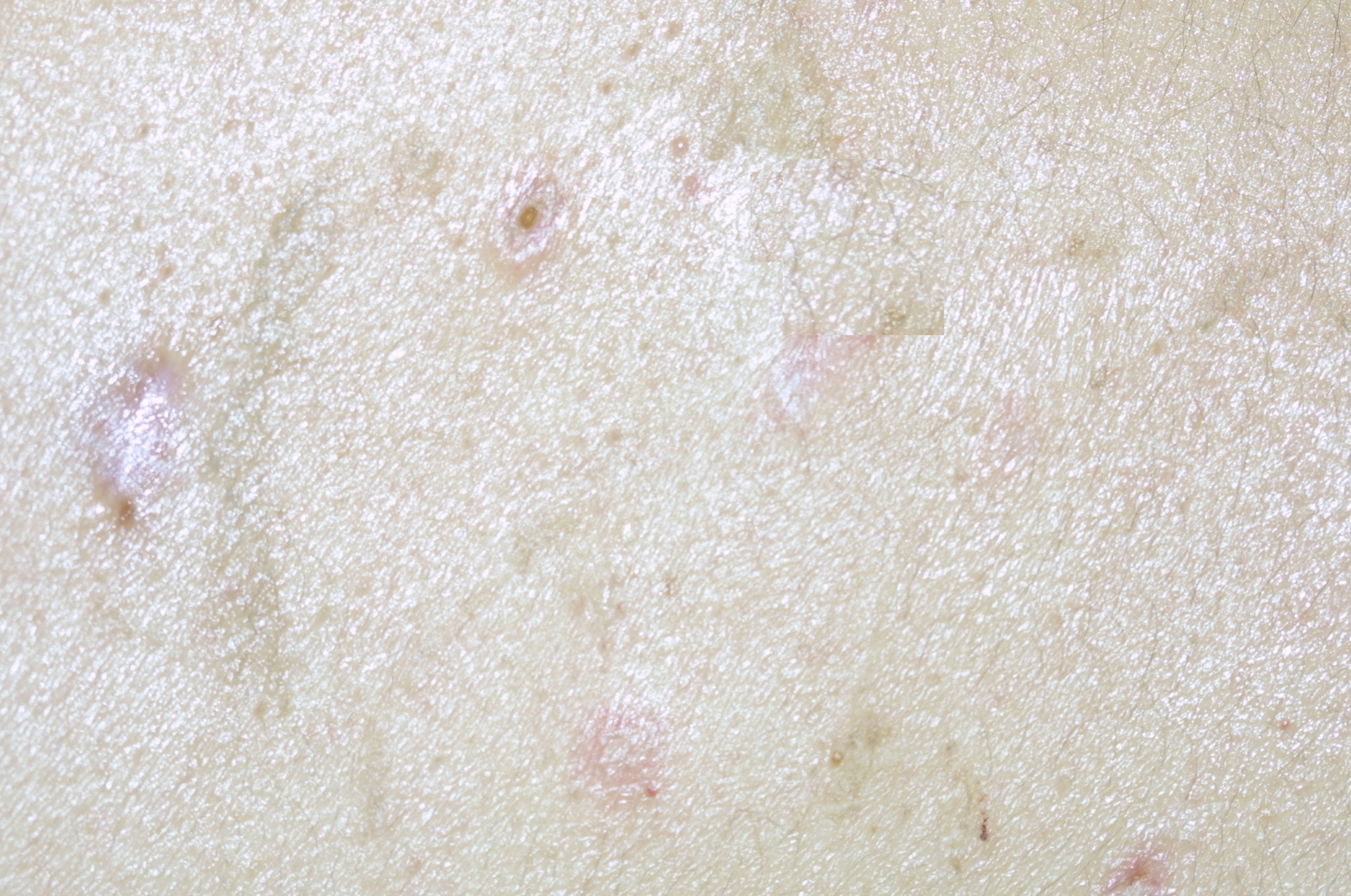 |
| Figure 1 | Figure 2 |
|---|
First, I must talk about the clinical findings of Degos Disease and specifically its typical skin findings. Degos Disease arises as erythematous papules; the differential diagnosis would include insect bites or a limited papular drug eruption (although the papules of Degos Disease are not typically itchy, folliculocentric, or initially generalized) [5]. Some patients state that these papules are accompanied by a burning sensation. These papules are most commonly 2-10 mm in diameter. The erythematous papules evolve into the prototypical, but not actually pathognomonic, white atrophic porcelain scars, which usually have a peripheral telangiectatic rim, the characteristic skin eruption of Degos Disease. DD can also manifest in the skin as erythematous ulcers, necrotic amputations, purpura, and cutaneous vasculitis [8].
It is important to recall that collagen vascular diseases (CVD) [9] (e.g., lupus [10], dermatomyositis [11], rheumatoid arthritis [12], progressive systemic sclerosis [13]) vasculitides such as Wegner Granulomatosis [14], thrombotic diseases such as thromboangiitis obliterans (Buerger disease [15]), and auto-immune diseases of the blood (e.g., antiphospholid syndrome [16]) can occasionally manifest initially with erythematous papules that evolve into white atropic scars, which look the same as the atrophic white scars of Degos Disease. A post-streptococcal vasculopathy with evolution to Degos Disease has been noted as well [17].
Second, I must discuss thrombosis and DD. Degos Disease is a vasculopathy or an endovasculitis. DD is not a frank vasculitis. It is not an immune complex type of vasculitis like leukocytoclastic vasculitis or the other types of vasculitis in the small, medium, and large vessel schema of vasculitis, but DD is an occlusive arteriopathy involving small-caliber vessels [18]. Broadly, Degos Disease manifests as a progressive, small- and medium-size arterial occlusive disease, leading to tissue infarction initially involving cutaneous blood vessels of the skin. Later DD can manifest as MAP in the internal organs in a minority of cases [5].
Third, I think it important to review the histological features of the skin in DD because it is important to understand that DD shares some of its features with CVD from which DD is distinct. The papules in Degos Disease can manifest with a primarily lymphocytic cell infiltrate associated with interstitial mucin deposition in a superficial and deep perivascular, periadnexal, and perineural pattern [19]. Eosinophils are almost never present. Occasionally, histiocytes contain engulfed nuclear debris. The mucin that is present in DD resembles the mucin found in specimens of the eruption of lupus [20]. In a fashion similar to lupus, DD can manifest with vacuolar alteration at the dermoepidermal junction [19]. The atrophic white papules of DD typically demonstrate wedge-shaped degeneration of collagen, sometimes along with mucin and vacuolar or interface change [19]. An interface dermatitis can be observed in DD but can be limited to the central portion of a specimen [19]. There can be squamatization of the dermoepidermal junction, melanin incontinence, epidermal atrophy, and dermal sclerosis, which resembles lichen sclerosis [5, 19]. One report notes that there can be necrobiosis of the collagen layer, which is reminiscent of granuloma annulare or necrobiosis lipodica [21]. Abundant acid mucopolysaccharides can manifest in the dermis, mimicking dermal mucinosis [20]. Extensive occlusive thrombotic microangiopathy with striking endothelial cell degeneration and sloughing can be present [1].
Direct immunofluorescence does not yield definitive findings in DD. Perivascular fibrin and complement can be found in specimens of DD [22]. An autopsy in a patient who manifested the clinical diagnosis of Degos disease demonstrated features suggestive of thromboangiitis of the Buerger type (i.e., bland infarcts of the small intestine and striking perforation of the jejunum) [23]. Observers have seen microaneurysms of the bulbar conjunctival vessels histologically and other eye changes [24, 25].
Magro (2011) [1] noted that in 3 of the 4 cases, “indirect immunofluorescent studies conducted on frozen tissue showed extensive deposits of C5b-9 within the cutaneous vasculature.” In the other case “the C5b-9 studies were conducted on paraffin-embedded tissue samples using an immunohistochemical technique whereby there was extensive deposition of C5b-9 in vessels, and there was marked extravascular C5b-9 deposition within the interstitium and amidst inflammatory cells.” In one case Magro (2011) noted that “indirect immunofluorescent studies conducted on frozen tissue showed extensive deposits of C5b-9 within the cutaneous vasculature.” There was extensive expression of MXA protein [MX1 (myxovirus resistance 1, encoding MXA)] in endothelial cells, the vessel walls, inflammatory cells, epidermal keratinocytes, and the perivascular interstitium.
Fourth, in DD there are changes and findings in the endothelial cells that resemble those of cells infected by a virus, not present in normal controls. Electron microscopy of the white papules of DD can demonstrate interwoven tubular structures within the endothelial cells, which have the quality of viral inclusions [26, 27]. Finding a virus in tissue of DD up until the report of Margo (2011) [1] had been a fruitless endeavor. Specifically, paramyxovirus was not found by polymerase chain reaction of DD tissue with viral inclusions [28]. Magro (2011) broke new ground by finding that in 3 of the 4 cases “there was positivity for parvovirus B19 RNA transcripts in the endothelium of the vasculature. Magro has suggested the B19 and DD have a relationship [29]. Other viruses, including herpes simplex and cytomegalovirus, were tested; the PCR results were negative.” However, as she notes “correlative antiviral serologic tests and or peripheral blood viral DNA tests” were not performed in her patients, making it uncertain if her findings of B19 RNA transcripts are an artifact, a side show or of no significance at all. As Magro (2011) rightly notes that “given the frequency of parvovirus B19 infection, one could argue that the identification of B19 RNA transcript expression in endothelial cells may not be of pathogenetic significance.” Finally, Magro (2011) notes that “[i]n an additional study, in situ B19 RNA expression was not identified in any skin samples in which there was no established role for parvovirus B19 in lesional propagation.” For the time being, it would seem, Degos Disease cannot be considered a viral-related condition.
Magro (2011) noted in her article that using electron microscopy in 2 cases “ultrastructural analysis showed extensive tubuloreticular structures in the epidermal keratinocytes and in endothelial cells throughout the dermis, consistent with exposure to IFN-α. The lining endothelial cells showed profound degenerative changes with detachment of the cells from the vessel lumens. The vascular basement membrane zones were reduplicated and showed collagen deposition.” The meaning of these structures remains uncertain.
Fifth and finally, no immune defect has ever been linked consistently to DD and no treatment that suppresses the immune system has consistently been shown to abate DD. In most instances of DD, no circulating immune complexes, anti-endothelial cell antibodies, or anti-cardiolipin antibodies can be identified [30, 31]. However, in some instances of DD, researchers have identified antiphospholipid antibodies of uncertain significance [32].
Medications (besides eculizumab) and toxic chemicals do not seem to induce Degos disease nor induce a remission of Degos Disease. A case of one patient with BAP who took prednisolone, azathioprine, and cyclosporine and later developed many additional lesions of DD has been noted [33]. One can only speculate if the drugs in this case were causative or not as DD often spreads in extent. Whereas IVIG and prednisone affect the course of many diseases with skin findings, they do not affect the course of Degos Disease [5, 34].
Whereas Magro (2011) notes that after using an anti–endothelial cell antibody assay in indirect immunofluorescent studies in one case “[t]here was conspicuous granular nuclear decoration in most of the endothelial cells with incubation with generic cutaneous endothelial cells followed by fluoresceinated human anti-IgG.” In this case “the IFN-α signature in the peripheral blood was very high.” In another case “an assay for anti-endothelial cell antibodies was also positive.” Whereas these findings are interesting because they are not consistent the significance of anti-endothelial cell antibodies remains uncertain. The fact that some cases of Degos Disease occur in lupus patients or look like lupus is well known and does not mean that Degos Disease in an autoimmune disease or that DD is lupus. As Magro (2011) notes “autoimmune disease, including scleroderma, antiphospholipid antibody syndrome, Behçet disease, SLE, and Henoch-Schönlein purpura, in association with “antibodies with endothelial cell specificity” has been noted. Thus, I will infer that currently the belief that Degos Disease is not an autoimmune disease or a disease of antibodies must stand.
Magro (2011) used Western Blot studies using generic endothelial cell lysates to analyze samples of her 4 study subjects. In one case, by using cutaneous endothelial cells as substrate, a distinct band of immunoreactivity corresponding to a molecular weight of 92 kDa was observed. A similar band of reactivity was not identified in normal control cases. Because these studies did not note positive results in all 4 subjects, the role of this band or protein is uncertain.
Previous criteria for Degos Disease must be amended
In 2007, I set forth 7 factors as possible criteria for defining DD, particularly the distinction of DD from lupus [6]. They were:
1. Unresponsiveness to therapy;2. Frequent presence of cellular virus-like inclusions;
3. Negative DIF findings;
4. Invariably fatal course in Systemic Degos Disease usually in 1-3 years from diagnosis;
5. Lack of photosensitivity;
6. Lack of facial lesions;
7. White atrophic papules whose histology is a sensitive rather than specific indication of DD.
I laid down these criteria with the understanding that physicians did not understand the basis or etiology of MAP. Previously, I had considered the relationship of DD to other diseases with similar features [36]. In 2011, these criteria must be reconsidered and must be at least amended if not abandoned altogether. The comparison that we must consider is no longer between lupus and DD but between DD and paroxysmal nocturnal hemoglobinuria.
Paroxysmal Nocturnal Hemoglobinuria and its relationship to Degos Disease
In May of 2011, in the face of Magro's report of April 2011 and data from a 2010 meeting at the Hospital for Special Surgery, New York City [35], pertaining to Degos Disease on the use of eculizumab to effective treat SDD/MAP, these criteria need to be reconsidered. This reconsideration is required because effective therapy now appears to exist for DD. In addition, we now understand that complement plays a role in the pathogenesis of Degos Disease. Further, the fact that eculizumab has been successfully deployed as a treatment of paroxysmal nocturnal hemoglobinuria (PNH) suggests a great deal about the pathogenesis of DD itself.
Any theory must explain all the available data and as yet no unified field therapy can be offered for most non-infectious disease because not all disease entities respond in an identical way to treatment. Whereas, this might imply that disease entities (e.g., psoriasis) are heterogeneous rather than homomorphous entities, it is also possible they are constellations of single entities. In this way, inflammatory or hematological diseases can be seen in a light similar to neoplastic diseases in that each patient is unique and the disease is an echo of its own individual genetic, infectious, and general environmental characteristics.
PNH is a rare, acquired, sometimes fatal hematological disease whose basis has been defined [37]. Like so many other diseases and like Degos Disease, PNH can arise on its own (“primary PNH”), in the context of other diseases, specifically bone marrow disorders such as aplastic anemia (“secondary PNH”), or be sub-clinical (i.e., PNH abnormalities which on flow cytometry lack signs of hemolysis) [38].
PNH can manifest with one, two, three, or more clinical findings. The three primary findings are: (1) complement-induced intravascular hemolytic anemia (most patients), (2) red urine (secondary to urinary hemoglobin, which occurs only in a fraction of cases), and (3) thrombosis (40% of patient and the key causes of death in PNH patients) [37]. A few PNH patients report abdominal pain, dysphagia, odynophagia, and/or erectile dysfunction.
PNH is a very rare disease with a prevalence of patients with the abnormal PNH clone in the region of 16 cases per million population. PNH appears to have an incidence of 1-2 per million population [39, 40]. It can be speculated that the prevalence and the incidence of Degos Disease might mirror that of PHN.
The thrombosis of PNH bears further explication. Unlike Degos Disease, which involves the small and medium vessels, the thrombosis of PNH involves larger blood vessels. In PNH, thrombosis most commonly manifests as deep vein thrombosis of the leg veins that results in pulmonary embolism [40]. Less commonly, PHN involves the hepatic vein (resulting in Budd-Chiari syndrome), hepatic portal vein, (resulting in portal vein thrombosis), the superior or inferior mesenteric veins (resulting in mesenteric ischemia), the cutaneous veins, and/or the cerebral veins (resulting in cerebral venous thrombosis) [41]. On a different note, 8 to 18 percent of deaths in PNH have been reported to have renal failure as at least a contributing factor [40].
PNH is the sole hemolytic anemia related to an acquired (rather than inherited) intrinsic defect in the cell membrane. Specifically, the defect involves deficiency of glycophosphatidylinositol leading to an absence of protective proteins in the membrane [42].
All blood cells (red blood cells, white blood cells, and platelets) need the enzyme phosphatidylinositol glycan A (PIGA) to manufacture glycosylphosphatidylinositol (GPI), a molecule that anchors proteins to their cell membranes. The biosynthesis of a glycosylphosphatidylinositol (GPI) structure requires the gene product PIGA to serve as an anchor for a group of membrane proteins [38, 40, 42]. The PIGA gene resides on the X chromosome. There is only one active copy in a cell (males have one X chromosome and females have one X chromosome because of the Lyon effect). In PNH, mutations in PIGA in bone marrow stem cells lead to GPI defects in all types of blood cells [43]. The development of PNH typically involves a hypoplastic bone marrow, somatic mutation restricted to the PIGA gene in the stem cell, and clonal expansion of the hematopoietic stem cell pool [44].
In PNH, blood cells are characterized by a total or partial lack of the GPI-anchored membrane proteins [43, 44]. Without this GPI structural apparatus, intravascular hemolysis occurs owing to the inability to regulate the lytic and cell-stimulatory activities of complement on the membrane surface of hematopoietic cells. The platelet dysfunction in PNH also contributes to the thrombosis [45]. Proteins including decay-accelerating factor (DAF or CD55) [46], which disrupts formation of C3 convertase [47], and protectin (CD59), which binds the membrane attack complex and prevents C9 from binding to the cells that anchor to GPI, protect the cells from complement mediated destruction [46, 47]. When CD55 and CD59 cannot bind to blood cells, owing to defective GPI, hemolysis, and thrombosis occur.
Other secondary mechanisms can affect the course of PNH. Esophageal spasm, erectile dysfunction, and abdominal pain relate to hemoglobin release during hemolysis because the hemoglobin binds with circulating nitric oxide (a smooth muscle relaxer) [48, 49]. The symptoms might be able to be ameliorated by administration of nitrates or sildenafil (Viagra), which improves the effect of nitric oxide on muscle cells.
The author believes that DD could be an acquired intrinsic defect in some endothelial or hematological gene and the protein or factor thereby expressed.
I would offer a literary passage from No Man's Land (1974-5) by Harold Pinter (p62) for how I imagine the devolution of Degos Disease or PNH occurs as a person wanders into a blind and deadly cul-de-sac. This cul-de-sac may lead to an acquired hematological disease like PNH or to a hematological or endothelial disease like Degos Disease.
He asked me the way to Bolsover street. I told him Bolsover street was in the middle of an intricate one-way system. It was a one way system easy enough to get into. The only trouble was that, once in, you couldn't get out. I told him I knew one or two people who'd been wandering up and down Bolsover street for years. The people who live there, their faces are grey, they're in a state of despair. I remember saying to him: This trip you've got in mind, drop it, it could prove fatal.
Heretofore, those who suffered from PNH or systemic Degos Disease (MAP) resided in a no man's land, which having been entered is all but impossible to escape. Pinter's play is make-believe and PNH and MAP are all too real, but all embody a murderous menace.
Degos Disease reconsidered as a Genetic and Hematological Disease
The fact that at least 10 cases of Degos Disease reported describe familial association underlies the possible genetic basis of Degos Disease [33, 50-53]. As Degos Disease is rare this familial incidence is far out of proportion to what would be expected from a disease that is not genetic. The Online Mendalian Inheritance in Man (OMIM) has given Degos disease a number – #602248 [54]. No gene has yet been cloned for DD and possible associated groups of genes might simply increase the odds ratio of developing Degos Disease, but the basis for understanding DD as a genetic disease exists.
Degos Disease might be a result of a genetic mutation and this mutation might be in the hematological system. The conceptualization of Degos Disease as a hematological disease is one that goes back decades. In 1993, Vázquez-Doval et al [55] noted 2 patients with DD. One showed an increase in plasminogen activator inhibitor-1 (PAI-1) activity and the second showed a decrease in platelet aggregation induced by adenosine diphosphate and adrenaline, but normal aggregation with collagen. Vázquez-Doval suggested “impairment of blood fibrinolytic activity and platelet aggregation may have pathogenic and therapeutic implications in malignant atrophic papulosis.” The interaction of complement, red blood cells, platelets, other blood factors, and the endothelial cells are complex and some dysfunction in these systems could underlie DD. The fact that GPI anchoring pathology can cause thrombosis would seem to enhance our understanding of thrombosis in DD.
The fact that DD is a hematological disease does not mean that all treatments for hematological diseases affect the course of DD. Some [56] have suggested that aspirin or dipyridamole can help reduce or abate the papules of DD, but this is anecdotal and this regimen has shown no affect on MAP [57].
Why some cases of DD remain confined to the skin and some are systemic is not certain. The two iterations of DD, one only in the skin and the other in the skin and internal organs, suggest that the interaction between the endothelial cells and the cells of the blood in the skin vasculature may be different from that of vessels in internal organs. Moreover, the vasculature of the skin may possess different molecular and dynamic properties in different body areas. That is, the endothelial cells in other areas of the body might be different than those on the face. Atrophic papulosis commonly occurs on the palms and soles and body. Whereas lupus commonly manifests on the face, I have not been able to find patients who have had lesions of Degos Disease on the face. I was unable to find any of the patients (about 20 patients who had attended the 2007 Degos conference at a meeting ancillary to the American College of Rheumatology Meeting or who are discussed in the literature) who have the lesions of Degos on their faces [58]. Simply put, we do not yet understand all the processes, cells, factors, or body regional differences involved with clotting.
Eculizumab
There is a monoclonal antibody named eculizumab that protects blood cells against immune destruction by inhibiting the complement system [59]. Eculizumab (Soliris) is a monoclonal antibody directed against the complement protein C5. Eculizumab blocks the cleavage of C5. Eculizumab exhibits its therapeutic activity by binding to the C5 complement protein, thereby blocking C5's cleavage by the C5 convertase [40, 42, 46, 59]. This blockage halts the process of complement-mediated cell destruction of red blood cells and platelets. Eculizumab has been shown to reduce the need for blood transfusion in patients with significant hemolysis who suffer from PNH. Eculizumab is effective in treating paroxysmal nocturnal hemoglobinuria and is FDA indicated for this use. Most patients with PNH who are treated with eculizumab will no longer require transfusions [40, 42, 46, 59].
Not all patients with PNH will respond in an effective fashion to eculizumab [40, 42, 46, 59]. Because PNH involves a complex array of factors that go beyond the PIGA gene and GPI protein and because clonal stem cell variants exist, this is to be expected. That is, only a proportion of patients with a specific PNH clone will benefit from eculizumab; this by extension would be true of treatment of Degos Disease with eculizumab. Some DD patients would respond to eculizumab and some DD patients would not.
As Magro (2011) noted, eculizumab has also been used to effectively treat other hematological diseases and forms of vascular thrombosis, including atypical hemolytic uremic syndrome [60], renal allograft rejection [61], and antiphospholipid syndrome [62]. Simply put, disabling the function of complement can abate a variety of hematological diseases.
In the publicly available data that was presented at HSS conference in 2010 on Degos Disease [35], in 3 patients with nearly fatal MAP, eculizumab worked immediately and was life saving with continued use. Whereas this series is anecdotal, it gives tremendous promise. The effectiveness of eculizumab emphasizes the idea that Degos Disease might not be an autoimmune disease, but instead, that Degos Disease is an endothelial, hematological, or complement-mediated disease. The optimal dosing for ezulizumab needs to be defined in Degos Disease but could be higher than that for PNH [1, 35]. Just as eculizumab does not work for every case of PNH it might not work for every case of MAP.
Placing Degos into a category of other diseases
In my article of 2009 on Degos [36] that was published in Dermatology Online Journal 15(1):10 entitled “Pairing and comparing nine diseases with Degos Disease (Malignant Atrophic Papulosis): An attempt to illustrate our understanding and direct future inquiry” I compared Degos Disease to a number of other entities. The reader is referred back to this article to consider these comparisons in detail but I will address them in short and relate them to new data.
Magro's (2011) molecular research suggests that DD is a dysregulated interferon-α response combined with membranolytic attack complex deposition that may contribute to the unique vascular changes. It is thereby considered a C5b-9/interferon-α-mediated endotheliopathy syndrome. However, it is possible that this is a secondary affect to some underlying genetic defect. Whereas some have suggested that DD is a variant of lupus, there seems to be little value in this comparison. Lupus occurs so much more frequently and differs so much from DD in response to treatment and findings. Eculizumab was originally investigated for use as a treatment for lupus, but these investigations and trials did not succeed at doses that work for PNH. It may be concluded for now that the idea that lupus and DD are in the same spectrum does not help define the etiology of DD. Finally, as rare diseases often relate to single (simple) genetic hits and are easier to define, one might imagine that the cause of DD will be defined before the cause of lupus is ascertained.
Conclusions
In conclusion, DD seems to be a hematological and/or endothelial disease that most likely relates to a genetic defect, perhaps an acquired (rather than inherited) intrinsic genetic defect like PNH. It is not clear if Degos Disease is a disease of the endothelial cell. Owing to the fact that endothelial cells differ in the face, the skin, the intestines, the CNS, and other internal organs it manifests differently and to different extents in these places and organ systems. It is not clear if Degos Disease is a disease of the blood and its cells and factors that manifests to different extents and in different fashions because of differences in the endothelial cells. It does seem clear that Degos Disease is not a disease of the immune system.
Therapy gives insight into the categorization of disease. In this way, we can see that Degos Disease is a hematological/endothelial disease and not an immune disease. Psoriasis does not respond to mycophenolate mofetil whereas atopic dermatitis does. Psoriasis responds to tumor necrosis factor α blockage whereas atopic dermatitis does not. Sneddon-Wilkinson disease (subcorneal pustular dermatosis) responds to dapsone and pustular psoriasis does not. Lupus can respond to IVIG, but psoriasis does not. If Degos Disease and PNH both respond to eculizumab then they perforce have something in common. If indeed Degos and anti-phospholipid syndrome do both respond to eculizumab then they might have more in common that we may initially have imagined.
Sometimes scientific serendipity and good luck allow us to see beyond the event horizon of a deadly disease. The work of Cynthia M. Magro, Jonathan C. Poe, Connie Kim, Lee Shapiro, Gerard Nuovo, Mary K. Crow, Yanick J. Crow, Christos C. Zouboulis, John Patrick Whelan, and the scientists at Alexion has served up something that inspires a sense of wonderment. That the 3 patients noted at the 2010 HSS conference who were dying of MAP (one of whom is patient #1 in Margo's 2011 article) were summoned back to life by the Lazarus clap of eculizumab, is stirring and moving.
In my decade of professional life as a dermatologist the riddles of nephrogenic systemic fibrosis (related to gadolinium), and hypereosinophilic syndrome, which is caused either by an interstitial chromosomal deletion on band 4q12 leading to the creation of the FIP1L1-PDGFRA fusion gene (F/P+ variant), or (2) increased interleukin (IL)-5 production by a clonally expanded T-cell population (lymphocytic variant) have been solved. So it will be with Degos Disease, if not now, then in the future. Like Moses and Martin Luther King we can see the promised land, if only distantly through a mirror darkly.
The paradigm of Kuhnian Normal Science posits that as much of the data must be explained to allow normal science research based on assumptions to occur. A practical reconciliation in the performance of actual scientific experiments of the Kuhnian dialectic with the kabuki play of the NIH grant process and the performance of the alchemy of science is beyond the scope of this paper. We as physicians and scientists now have a basis for normal scientific research to proceed to unravel Degos Disease. Margo's work paves the way of the exercise of Normal Science to unravel the full basis of Degos Disease. A hopeless parsing of all disease that occurs in the skin into TH-1 and TH-2 responses can be abandoned. Out of the darkness and death of MAP comes a new hope and it had to be that way. If the right hand of Degos Disease was dark, the left of this darkness is light. Let us go forward armed two handed to grapple with MAP – one hand holding the science based on the idea that DD involves complement and the other hand holding eculizumab.
Conflict-of-interest disclosure: The author has received honoraria funding from Alexion Pharmaceuticals Inc. worth less than $3000.
References
1. Magro CM, Poe JC, Kim C, Shapiro L, Nuovo G, Crow MK, Crow YJ. Degos disease: a C5b-9/interferon-α-mediated endotheliopathy syndrome. Am J Clin Pathol 2011;135:599-610. [PubMed]2. Degos R., J. Delort, R. Tricot (1942). Dermatite papulosqameuse atrophiante. Bull. Soc. Fr. Derm. Syph. 49, 148-150.
3. Degos R. Malignant atrophic papulosis. Br J Dermatol 1979; 100: 21-35. [PubMed]
4. Kohlmeier W. Multiple Hautnelnosen bei Thromboangiitis obliterans. Arch Dermatol Syphilol 1941; 181: 783-4.
5. Scheinfeld N. Malignant atrophic papulosis. Clin Exp Dermatol. 2007 Sep;32(5):483-7. [PubMed]
6. Scheinfeld N. Degos' disease is probably a distinct entity: a review of clinical & laboratory evidence. J Am Acad Dermatol. 2005;52(2):375-6; author reply 376-8. [PubMed]
7. Fernández-Pérez ER, Grabscheid E, Scheinfeld NS. A case of systemic malignant atrophic papulosis (Köhlmeier-Degos' disease). J Natl Med Assoc. 2005 Mar;97(3):421-5. [PubMed]
8. Scheinfeld NS, Degos Disease, eMedicine, (Accessed June 26, 2011) http://emedicine.medscape.com/article/1087180-overview
9. Ortiz A, Ceccato F, Albertengo A, Roverano S, Iribas J, Paira S. Degos cutaneous disease with features of connective tissue disease. J Clin Rheumatol. Apr 2010;16(3):132-4.
10. Mutizwa MM, Tang MB, Ng SK. Prominent Degos-like skin lesions in a patient with chronic cutaneous lupus erythematosus. Dermatol Online J. 2010 Jul 15;16(7):5. [PubMed]
11. Tsao H, Busam K, Barnhill RL, Haynes HA. Lesions resembling malignant atrophic papulosis in a patient with dermatomyositis. J Am Acad Dermatol. 1997 Feb;36(2 Pt 2):317-9. [PubMed]
12. Demitsu T, Kakurai M, Murata S, Kiyosawa T, Yaoita H. Degos' disease associated with rheumatoid arthritis. J Dermatol. 1997 Jul;24(7):488-90. [PubMed]
13. Liu CM, Harris RM, Hansen CD. Lesions resembling malignant atrophic papulosis in a patient with progressive systemic sclerosis. Cutis. 2005 Feb;75(2):101-4. [PubMed]
14. Frank H, Metz J, Müller E. Papulosis atrophicans maligna (Degos). A form of endangitis obliterans. Hautarzt. 1974 Sep;25(9):432-7. [PubMed]
15. Guhl G, Diaz-Ley B, Delgado Y, Dauden E, Fraga J, Garcia-Diez A. Wegener's granulomatosis: a new entity in the growing differential diagnosis of Degos' disease. Clin Exp Dermatol. Jul 2009;34(5):e1-3. [PubMed]
16. Grattan CE, Burton JL. Antiphospholipid syndrome and cutaneous vasoocclusive disorders. Semin Dermatol. 1991 Sep;10(3):152-9. [PubMed]
17. Pati S, Muley SA, Grill MF, Coons S, Walker R. Post-streptococcal vasculopathy with evolution to Degos' disease. J Neurol Sci. 2011 Jan 15;300(1-2):157-9. [PubMed]
18. Sibillat M, Avril MF, Charpentier P, Offret H, Bloch-Michel E. Malignant atrophic papulosis (Degos' disease): clinical review. Apropos of a case. J Fr Ophtalmol. 1986;9(4):299-304. [PubMed]
19. Harvell JD, Williford PL, White WL. Benign cutaneous Degos' disease: a case report with emphasis on histopathology as papules chronologically evolve. Am J Dermatopathol. 2001 Apr;23(2):116-23. [PubMed]
20. Zambal Z. Cutaneous deposits in alopecia mucinosa, Degos' papulosis atrophicans maligna and Buschke's scleredema. Arch Klin Exp Dermatol. 1970;237(1):105-8. German. [PubMed]
21. Yoshikawa H, Maruta T, Yokoji H, Takamori M, Yachie A, Torii Y. Degos' disease: radiological and immunological aspects. Acta Neurol Scand. 1996 Nov;94(5):353-6. [PubMed]
22. Crowson AN, Magro CM, Usmani A, McNutt NS. Immunoglobulin A-associated lymphocytic vasculopathy: a clinicopathologic study of eight patients. J Cutan Pathol. 2002 Nov;29(10):596-601. [PubMed]
23. Stejskalová A, Stanová M, Vosmík F. Malignant atrophic papulosis (Degos' syndrome). Sb Lek. 1990 Jan;92(1):1-5. [PubMed]
24. Ophthalmic changes of Degos' disease (malignant atrophic papulosis). Lee DA, Su WP, Liesegang TJ. Ophthalmology. 1984 Mar;91(3):295-9. [PubMed]
25. Egan R, Lessell S. Posterior subcapsular cataract in Degos disease. Am J Ophthalmol. Jun 2000;129(6):806-7. [PubMed]
26. Olmos L, Hunziker N, Laugier P. Microcylinders of endoplasmic reticulum in histiocytes in patients suffering from Degos' syndrome and dermatomyositis. Br J Dermatol. 1979 Feb;100(2):137-45.[PubMed]
27. Bioulac P, Doutre MS, Beylot C. Degos' malignant atrophic papulosis. Ultrastructural study of a new case. Ann Anat Pathol (Paris). 1980;25(2):111-24. [PubMed]
28. D'Avino M, Lo Schiavo A, Baroni A, Buommino E, Ruocco E.Degos' disease: a case with cutaneous lesions only: absence of paramyxovirus by PCR. Dermatology. 2000;201(3):278-9. [PubMed]
29. Dyrsen ME, Iwenofu OH, Nuovo G, Magro CM. Parvovirus B19-associated catastrophic endothelialitis with a Degos-like presentation. J Cutan Pathol. 2008 Oct;35 Suppl 1:20-5. [PubMed]
30. Levine SR, Welch KM. The spectrum of neurologic disease associated with antiphospholipid antibodies. Lupus anticoagulants and anticardiolipin antibodies. Arch Neurol. 1987 Aug;44(8):876-83. [PubMed]
31. Hohwy T, Jensen MG, Tøttrup A, Steiniche T, Fogh K. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leinden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86(3):245-7. [PubMed]
32. Asherson RA, Cervera R.Antiphospholipid syndrome. J Invest Dermatol. 1993 Jan;100(1):21S-27S. [PubMed]
33. Powell J, Bordea C, Wojnarowska F, Farrell AM, Morris PJ. Benign familial Degos disease worsening during immunosuppression. Br J Dermatol. 1999 Sep;141(3):524-7. [PubMed]
34. De Breucker S, Vandergheynst F, Decaux G.Inefficacy of intravenous immunoglobulins and infliximab in Degos' disease. Acta Clin Belg. 2008 Mar-Apr;63(2):99-102. [PubMed]
35. Magro C, Shapiro L, Johnson B, Salmon J, Scheinfeld N, Whelan P. Rheumatology Grand Rounds Hospital for Special Surgery (New York City) Degos Disease. Feb 10, 2010.
36. Scheinfeld N. Pairing and comparing nine diseases with Degos Disease(Malignant Atrophic Papulosis): an attempt to illustrate our understanding and direct future inquiry. Dermatol Online J. 2009;15(1):10. [PubMed]
37. Savage WJ, Brodsky RA.New insights into paroxysmal nocturnal hemoglobinuria. Hematology. 2007 Oct;12(5):371-6. [PubMed]
38. Nagarajan S, Brodsky RA, Young NS, Medof ME.Genetic defects underlying paroxysmal nocturnal hemoglobinuria that arises out of aplastic anemia. Blood. 1995 Dec 15;86(12):4656-61. [PubMed]
39. Gulbis B, Eleftheriou A, Angastiniotis M, Ball S, Surrallés J, Castella M, Heimpel H, Hill A, Corrons JL. Epidemiology of rare anaemias in Europe. Adv Exp Med Biol. 2010;686:375-96. Review. [PubMed]
40. Hillmen P. The role of complement inhibition in PNH. Hematology. Am Soc Hematol Educ Program. 2008:116-23. [PubMed]
41. Yin DL, Liu LX, Zhang SG, Tian LT, Lu ZY, Jiang HC. Portal hypertension resulted from paroxysmal nocturnal hemoglobinuria: a case report and review of literature. Int J Hematol. 2009 Apr;89(3):302-4. Epub 2009 Mar 26. [PubMed]
42. Kelly R, Richards S, Hillmen P, Hill A. The pathophysiology of paroxysmal nocturnal hemoglobinuria and treatment with eculizumab. Ther Clin Risk Manag. 2009;5:911-21. [PubMed]
43. Almeida A, Layton M, Karadimitris A. Inherited glycosylphosphatidyl inositol deficiency: a treatable CDG. Biochim Biophys Acta. 2009 Sep;1792(9):874-80. Epub 2009 Jan 9. Review. [PubMed]
44. Bessler M, Hiken J. The pathophysiology of disease in patients with paroxysmal nocturnal hemoglobinuria. Hematology Am Soc Hematol Educ Program. 2008:104-10. [PubMed]
45. Weitz IC.Thrombosis in patients with paroxysmal nocturnal hemoglobinuria. Semin Thromb Hemost. 2011 Apr;37(3):315-21. [PubMed]
46. Parker C. Eculizumab for paroxysmal nocturnal haemoglobinuria. Lancet. 2009 Feb 28;373(9665):759-67. [PubMed]
47. Risitano AM, Perna F, Selleri C.Achievements and Limitations of Complement Inhibition by Eculizumab in Paroxysmal Nocturnal Hemoglobinuria: The Role of Complement Component 3. Mini Rev Med Chem. 2011 May 11. [PubMed]
48. Hill A, Rother RP, Wang X, Morris SM Jr, Quinn-Senger K, Kelly R, Richards SJ, Bessler M, Bell L, Hillmen P, Gladwin MT. Effect of eculizumab on haemolysis-associated nitric oxide depletion, dyspnoea, and measures of pulmonary hypertension in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2010 May;149(3):414-25. [PubMed]
49. Hill A, Richards SJ, Hillmen P. Recent developments in the understanding and management of paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2007 May;137(3):181-92. [PubMed]
50. Katz SK, Mudd LJ, Roenigk HH Jr. Malignant atrophic papulosis (Degos' disease) involving three generations of a family. J Am Acad Dermatol. 1997 Sep;37(3 Pt 1):480-4. No abstract available. [PubMed]
51. Habbema L, Kisch LS, Starink TM. Familial malignant atrophic papulosis (Degos' disease)--additional evidence for heredity and a benign course. Br J Dermatol. 1986 Jan;114(1):134-5. [PubMed]
52. Kisch LS, Bruynzeel DP. Six cases of malignant atrophic papulosis (Degos' disease) occurring in one family. Br J Dermatol. 1984 Oct;111(4):469-71. [PubMed]
53. Moulin G, Barrut D, Franc MP, Pierson A. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111(2):149-55. [PubMed]
54. Malignant Atrophic Papulosis http://www.ncbi.nlm.nih.gov/omim/602248
55. Vázquez-Doval FJ, Ruiz de Erenchun F, Páramo JA, Quintanilla E. Malignant atrophic papulosis. A report of two cases with altered fibrinolysis and platelet function. Clin Exp Dermatol. 1993 Sep;18(5):441-4. [PubMed]
56. Drucker CR. Malignant atrophic papulosis: response to antiplatelet therapy. Dermatologica. 1990;180(2):90-2. [PubMed]
57. Melnik B, Hariry H, Vakilzadeh F, Gropp C, Sitzer G. Malignant atrophic papulosis (Köhlmeier-Degos disease). Failure to respond to interferon alpha-2a, pentoxifylline and aspirin]. Hautarzt. 2002 Sep;53(9):618-21. [PubMed]
58. http://www.degosdisease.com/events/2007_11_09-program (Accessed December 1, 2007).
59. Schrezenmeier H, Höchsmann B. Eculizumab opens a new era of treatment for paroxysmal nocturnal hemoglobinuria. Expert Rev Hematol. 2009 Feb;2(1):7-16. [PubMed]
60. Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. 2009 Jan 29;360(5):544-6. [PubMed]
61. Chandran S, Baxter-Lowe L, Olson JL, Tomlanovich SJ, Webber A. Eculizumab for the treatment of de novo thrombotic microangiopathy post simultaneous pancreas-kidney transplantation-a case report. Transplant Proc. 2011 Jun;43(5):2097-101. [PubMed]
62. Lonze BE, Singer AL, Montgomery RA. Eculizumab and renal transplantation in a patient with CAPS. N Engl J Med. 2010 May 6;362(18):1744-5. [PubMed]
© 2011 Dermatology Online Journal