Vascular Ehlers-Danlos syndrome: A case with fatal outcome
Published Web Location
https://doi.org/10.5070/D30997g30qMain Content
Vascular Ehlers-Danlos syndrome: A case with fatal outcome
Paulo Morais1,2, Alberto Mota1,2, Catarina Eloy3, José Manuel Lopes2,3,4, Fátima Torres5, Aida Palmeiro5, Purificação Tavares5, Filomena Azevedo1
Dermatology Online Journal 17 (4): 1
1. Department of Dermatovenereology, Hospital S. João, Porto, Portugal2. Faculty of Medicine, University of Porto, Porto, Portugal
3. Department of Pathology, Hospital S. João, Porto, Portugal
4. Institute of Molecular Pathology and Immunology of the University of Porto, Porto, Portugal
5. CGC Genetics, Porto, Portugal
Abstract
A 13-year-old boy, born prematurely and hypotonic, from non-consanguineous healthy parents, was referred to our department because of easy bruising. A slightly extensible, thin and translucent skin, associated with dysmorphic facies, acrogeria, multiple ecchymoses, hypermobility of the small joints, dorsal kyphosis, genu valgum, flat feet, elongated upper limbs, and low muscle tone were all evident. A history of learning disability and bilateral inguinal hernia was present. Blood and imaging studies were unremarkable. A skin biopsy disclosed an unremarkable dermis; electron microscopy showed abnormalities in the diameter, contour, and shape of collagen fibrils/fibers. Genetic analysis revealed heterozygosity for a novel mutation in COL3A1 gene (c.3527G>A), confirming the diagnosis of vascular Ehlers-Danlos syndrome (VEDS). The patient died at 15 years of age because of aortic dissection. Vascular Ehlers-Danlos syndrome is a rare, life-threatening, autosomal dominant variant of EDS, resulting from mutations in COL3A1 gene. Affected individuals are prone to serious and potentially fatal complications, especially vascular, intestinal, and uterine ruptures. Delay in diagnosis is common, even when the clinical presentation is typical. Therefore, dermatologists should be familiar with VEDS features because the skin findings may be the first signs. Early diagnosis will improve management of visceral complications and allow early genetic counseling.
Introduction
Ehlers-Danlos syndrome (EDS) is a heterogeneous group of inherited connective tissue disorders characterized by skin hyperextensibility, joint hypermobility, and tissue fragility [1, 2, 3]. The prevalence is estimated to be approximately 1 in 5000 births, with no racial predisposition [1]. The first description of the disorder was probably around 1682, but it was not until the early 1900s that the typical features of the syndrome were reported by Ehlers [4] and Danlos [5].
The Villefranche classification [6], introduced in 1997, differentiates six types of EDS, including vascular EDS (VEDS; EDS type IV; OMIM #130050). Vascular Ehlers-Danlos syndrome was first described by Sack in 1936 [7] and later was recognized as a distinct clinical entity by Barabas in 1967 [8]. Its biochemical basis was first identified in 1975 by Pope et al [9]. Vascular Ehlers-Danlos syndrome accounts for 4 percent of all EDS cases and is the most severe subtype, often causing premature death owing to arterial, bowel, or uterine rupture [10-16].
Case report
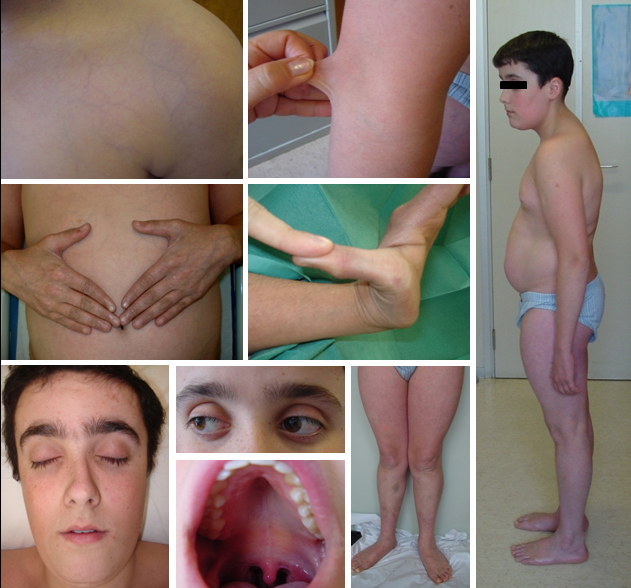 |
| Figure 1 |
|---|
| Figure 1. Clinical appearance of our patient. Note the translucent skin with visible underlying vasculature, slightly extensible skin, acrogeria, small joint hypermobility, characteristic facies, ecchymosis on the legs, genu valgum, elongated upper extremities, and dorsal kyphosis. |
A 13-year-old boy was referred to our department because of easy bruising. He was born prematurely at 35 weeks and 6 days via spontaneous vaginal delivery to non-consanguineous healthy parents. Except for hypotonia at birth, the remaining perinatal and antenatal history was unremarkable. His younger brother was asymptomatic and there was no family history of premature sudden death or relevant diseases. He had a history of bruising and hematomas after trivial injuries and bilateral inguinal hernia repair at the age of 9. Because of learning disabilities and an abnormal physical phenotype he had been studied for a possible fragile X syndrome (FXS). However, genetic analysis of the FMR1 gene was normal. In addition, a complete but unremarkable study for coagulation disorders had been performed. Physical examination revealed a thin, smooth, soft, translucent and slightly extensible skin, with highly visible subcutaneous vessels. An elongated face with pinched nose, thin upper lip, high-arched palate, small chin, prominent bulging eyes, and long eyelashes was also evident. In addition, hyperpigmented, speckled, prematurely aged skin of the extremities was noted. Other associated abnormalities were hypermobility, particularly of the small joints, dorsal kyphosis, genu valgum, flat feet, long upper limbs, low muscle tone, and multiple ecchymoses, especially on the legs (Figure 1). This clinical presentation suggested a disease of the connective tissue as the underlying cause. In fact, he had no dysmorphic features consistent with Marfan syndrome, but clear signs of Ehlers-Danlos syndrome, particularly its vascular variant.
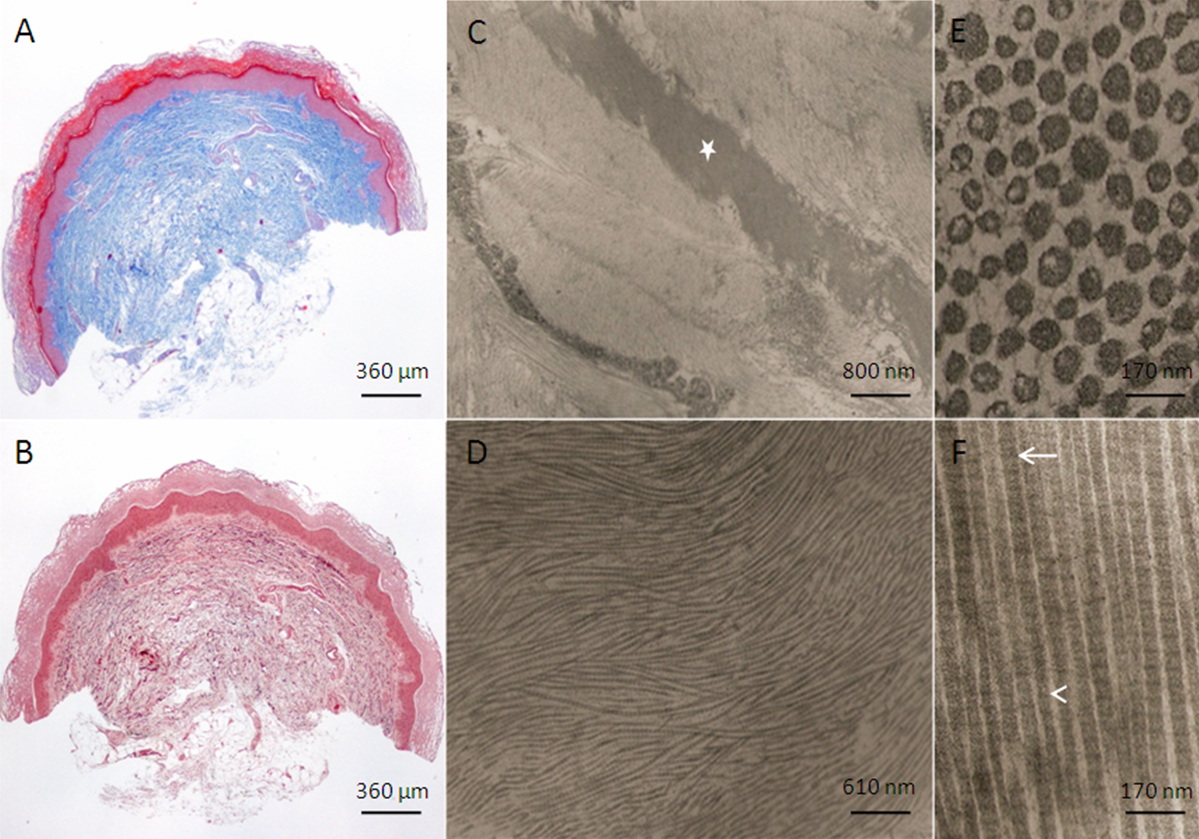 |
| Figure 2 |
|---|
| Figure 2. Trichromic (A) and orcein (B) features of the skin biopsy, in Ehlers-Danlos syndrome. Low power ultrastructural features of the reticular dermis (C) with elastic fibers (C – star) and undulating (D), transverse (E) and longitudinal (F) aspects of the collagen fibers (uranyl acetate and lead citrate). |
Complete blood cell count, coagulation studies, echocardiography, and abdominal ultrasonography were all normal. Skin biopsy disclosed unremarkable dermis, 1 mm largest thickness (Figure 2a), without apparent alterations of the elastic fiber component (Figure 2b). Ultrathin sections from the reticular dermis (Figure 2c) allowed longitudinal and transverse evaluation of collagen fibers. In this case it was possible to observe fibrils with a reduced diameter (32 nm) in up to 18 percent of those within packed fibers (mean diameter: 39 nm), some of them with irregular contours and internal electron-lucent aspects (Figure 2e). Some fibers had alterations of the collagen fibrillar curvature (Figure 2d) and discrete variation of diameter (Figure 2f, arrow, arrow point). Collagen periodicity was 65 nm (Figure 2f). Elastic fibers (Figure 2c, star) were apparently normal.
 |
| Figure 3 |
|---|
| Figure 3. Contrast-enhanced axial CT image during arterial phase demonstrates an intimal flap (arrow) that separates the two channels in the descending aorta diagnostic of a Stanford type A dissection. Note the true (t) and the false (f) lumen, and the presence of hemomediastinum (asterisk). |
Biochemical analysis of cultured skin fibroblasts showed normal type I, III and V collagen. However, DNA sequence analysis of the entire coding region of the COL3A1 gene, including intron-exon boundaries, revealed a heterozygous state for a novel point mutation in exon 49, changing G to A at base pair 3527 of the collagen α1(III) cDNA (c.3527G>A). This single-nucleotide substitution results in the shift of glycine with aspartic acid at amino acid position 1176 of the α1(III)-chain (p.Gly1176Asp). These findings were in agreement with the clinical diagnosis. Unfortunately, the patient died at 15 years of age owing to a sudden aortic dissection type A (Stanford classification), before the results of the molecular genetic studies were available (Figure 3). The molecular genetic analysis of the proband’s parents was negative, suggesting a spontaneous mutation.
Discussion
Vascular Ehlers-Danlos syndrome is a distinct form of EDS caused by mutations in COL3A1, a gene with 51 exons located in the long arm of chromosome 2 (2q32.2). This gene encodes the pro-α1 chain of type III collagen, a major component of distensible tissues such as skin, artery walls, and hollow viscera [11, 12, 13, 14]. The disease is transmitted as an autosomal dominant trait, however, the incidence of mutations is high and sporadic cases account for half of reported cases [10-15]. Because no genetic alterations were found in the genetic study of the COL3A1 gene in the parents, it is reasonable to assert that the detected mutation was not inherited but de novo. As far as we know, the c.3527G>A mutation had not been previously reported in the medical literature. A wide spectrum of COL3A1 mutations has been identified, the majority (approximately two-thirds) being point mutations leading to substitutions for the obligatory glycine in the triple helical region of the collagen molecule [15, 17]. According to Persikov et al [18] any glycine replacement in the collagen triple helix will cause clinical disease and this was the case of our patient, in which glycine was replaced by aspartic acid (p.Gly1176Asp). Most mutations are “private,” meaning that they are found in a specific family and with no correlation between genotype and phenotype [1, 13, 15]. Nevertheless, some researchers have tried to find a possible linkage between the “acrogeric” phenotype and the presence of mutations that alter sequences in the carboxyl-terminal end of the type III collagen triple helix [19], as observed in our patient. This is not the opinion of most authors at present.
Clinical diagnosis of VEDS is based on four major criteria (Table 1) [6, 11, 12, 13, 15, 16]: 1) a characteristic facial appearance, including elongated, triangular, and emaciated face with prominent staring eyes, often with periorbital pigmentation and fine telangiectasias on the eyelids, thin lips (particularly the upper lip whose edges are undefined) lobeless ears, small chin, and thin scalp hair; 2) a thin, pale and translucent skin with highly visible subcutaneous vessels, especially over the chest, abdomen and extremities, that unlike other types of EDS is not hyperelastic; 3) extensive bruising; and 4) arterial, digestive, and/or obstetrical complications. Although the presence of two of these criteria is highly indicative of VEDS, further testing is strongly recommended to confirm the diagnosis, namely biochemical assays (sodium dodecyl-sulfate polyacrylamide gel electrophoresis [SDS-PAGE]) of the type III collagen molecules synthesis in cultured fibroblasts and/or the identification of a mutation in the COL3A1 gene [1, 6, 11-16, 20]. Our study demonstrates that the standard biochemical testing may produce false negative results and that a search for a COL3A1 mutation can be diagnostic because it can identify additional cases that may be missed by SDS-PAGE. On the other hand, despite providing diagnostic certainty if a mutation is detected, the genetic study has a sensitivity of only 61 percent [15]. The ultrastructural abnormalities that we found are similar to those reported in the literature [21, 22] and, although they do not allow a specific classification of EDS subtype, suggest a disturbed fibrillogenesis and further give evidence that EDS is a disease of fibroblasts leading to changes of collagen [21].
As observed in our patient, hypermobility of large joints, characteristic of other types of EDS, is an uncommon finding in patients with VEDS [1, 6, 12, 13, 20]. In fact, he suffered mainly from hypermobility of the small joints of the hands and feet. Neonates may present with clubfoot and/or congenital dislocation of the hips [15]. In childhood, inguinal hernia, pneumothorax, and recurrent joint dislocation are common. In women suffering from VEDS, pregnancy can increase the likelihood of a uterine or vascular rupture. The reported maternal mortality rate is around 12 percent [15].
Vascular Ehlers-Danlos syndrome can be life-threatening because of weak arteries, bowel, or uterus, which can lead to spontaneous ruptures. These complications, normally rare in childhood, affect 25 percent of VEDS patients before the age of 20, and more than 80 percent by the age of 40 [15]. The mean life expectancy is between 48 and 54 years [15, 20]. Arterial dissection or rupture with uncontrolled bleeding is the main cause of death [15, 23], which unfortunately occurred in our patient.
Many health professionals are not familiar with VEDS, which is sometimes mistaken for coagulation disorders, Silverman syndrome, or physical abuse, especially in children. Vascular Ehlers-Danlos syndrome can also be confused with other types of EDS, Marfan syndrome, Loeys-Dietz syndrome or arterial tortuosity syndrome, in adulthood [11, 13]. Our patient was first thought to have an FXS because, besides learning problems, typical features of that disorder were present, namely elongated face, high-arched palate, flat feet, hyperextensible finger joints, and low muscle tone. As discussed above, these findings are also present in VEDS, the confirmed diagnosis of our patient.
There is still no effective treatment to prevent the complications associated with VEDS. Nonetheless, management by an experienced multidisciplinary team and implementation of some preventive measures, including the avoidance of contact sports, isometric exercises, and situations or drugs likely to raise blood pressure, should help to reduce the risk of complications [2]. Artery puncture, surgery, arteriograms, and endoscopies are contraindicated, allowed only in life-threatening emergencies [11, 13]. Psychological treatment and support of patients and their families are essential [2]. Prenatal diagnosis can be considered in families in which the mutation is known. However, choriocentesis and amniocentesis may be hazardous for the pregnant woman [11].
Although gene therapy is an important option for the treatment of many genetic disorders, the defective gene cannot be replaced in dominantly inherited diseases such as VEDS [14]. The recently developed RNA interference (RNAi) technology, which enables gene posttranscriptional expression silencing, may be applicable. In fact, Watanabe et al [24] showed the in vitro feasibility of a cellular therapy restoring collagen III synthesis in patients’ fibroblasts, using RNAi mediated inhibition of the COL3A1 mutation allele and transcriptional activation of the normal allele. According to these authors, this technique can provide a future cure for individuals with VEDS [14]. Likewise, a recent multicenter, randomised trial (NCT00190411) tested the protective effect of the beta-blocker, celiprolol on cardiovascular events of VEDS patients [25]. Celiprolol effectively reduced the incidence of arterial rupture or dissection by three times, over a median follow-up of 47 months, and might be, according to the authors, the treatment of choice to prevent major complications in patients with VEDS.
Clinical awareness and timely diagnosis of VEDS are still inadequate, even when the clinical picture is typical, resulting in unnecessary or inappropriate treatment and management [16, 17]. Therefore, the disease is often diagnosed only after life-threatening complications or death [15, 17, 20]. Although no specific treatment exists for VEDS, knowledge of the diagnosis is crucial, given its implications in daily life activities, pregnancy, surgical, and anesthetic decisions, genetic counseling, and management of major complications [15]. The use of a medical alert bracelet with the diagnosis “Vascular Ehlers-Danlos Syndrome” is highly recommended. Finally, if this proves feasible, RNAi technology based therapy and beta-blockers are promising options for a real improvement of VEDS patients’ prognosis and survival in the future.
Acknowledgment: The collaboration of the Center for Medical Genetics (Ghent, Belgium) with the biochemical and molecular studies is acknowledged. The authors are also grateful to proband’s family for their interest and support in the investigations
References
1. Steinmann B, Royce P, Superti-Furga A. The Ehlers-Danlos syndrome. In: Royce P, Steinmann B, eds. Connective tissue and its heritable disorders: molecular, genetic, and medical aspects, 2nd edn. New York; Wiley-Liss, Inc., 2002; 431-523.2. Callewaert B, Malfait F, Loeys B, De Paepe A. Ehlers-Danlos syndromes and Marfan syndrome. Best Pract Res Clin Rheumatol 2008; 22: 165-189. [PubMed]
3. Parapia LA, Jackson C. Ehlers-Danlos syndrome – a historical review. Brit J Haematol 2008; 141: 32-35. [PubMed]
4. Ehlers E. Cutis laxa, neigung zu haemorrhagien in der haut, lockerung meherer artikulationen. Dermatol Z 1901; 8: 173-174.
5. Danlos M. Un cas de cutis laxa avec tumeurs par contusion chronique des coudes et des genoux (xanthome juvénile pseudo-diabétique de MM. Hallopeau et Macé de Lépinay). Bull Soc Franc Dermatol Syph 1908; 19: 70-72.
6. Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet 1998; 77: 31-37. [PubMed]
7. Sack G. Status dysvascularis, ein fall von besonder zerreisslichkeit der blutgefasse. Dtsch Arch Klin Med 1936; 178: 663-669.
8. Barabas AP. Heterogeneity of the Ehlers-Danlos syndrome: description of three clinical types and a hypothesis to explain the basic defect(s). Br Med J 1967; 2: 612-613. [PubMed]
9. Pope FM, Martin GR, Lichtenstein JR, et al. Patients with Ehlers-Danlos syndrome lack type III collagen. Proc Natl Acad Sci USA 1975; 72: 1314-1316. [PubMed]
10. Barabas A. Vascular complications with Ehlers-Danlos syndrome with special reference to the “arterial type” of Sack’s syndrome. J Cardiovasc Surg (Torino) 1972; 13: 160-167. [PubMed]
11. Germain DP. Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis 2007; 2: 32. [PubMed]
12. Perdu J, Boutouyrie P, Lahlou-Laforêt K, et al. Syndrome d’Ehlers-Danlos vasculaire. Presse Med 2006; 35: 1864-1875. [PubMed]
13. Germain DP, Herrera-Guzman Y. Vascular Ehlers-Danlos syndrome. Ann Genet 2004; 47: 1-9. [PubMed]
14. Watanabe A, Shimada T. The vascular type of Ehlers-Danlos syndrome. J Nippon Med Sch 2008; 75: 254-261. [PubMed]
15. Pepin M, Schwarze U, Superti-Furga A, et al. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000; 342: 673-680. [PubMed]
16. Germain DP. Clinical and genetic features of vascular Ehlers-Danlos syndrome. Ann Vasc Surg 2002; 16: 391-397. [PubMed]
17. Watanabe A, Kosho T, Wada T, et al. Genetic aspects of the vascular type of Ehlers-Danlos syndrome (vEDS, EDSIV) in Japan. Circ J 2007; 71: 261-265. [PubMed]
18. Persikov AV, Pillitteri RJ, Amin P, Schwarze U, Byers PH, Brodsky B. Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders. Hum Mutat 2004; 24: 330-337. [PubMed]
19. Johnson PH, Richards AJ, Lloyd JC, Pope FM, Hopkinson DA. Efficient strategy for the detection of mutations in acrogeric Ehlers-Danlos syndrome type IV. Hum Mutat 1995; 6: 336-342. [PubMed]
20. Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg 2005; 42: 98-106. [PubMed]
21. Vitellaro-Zuccarello L, Cheli F, Esposito R, Bairati A. Ultrastructural study of the dermis in a case of type IV Ehlers-Danlos syndrome. J Submicrosc Cytol 1985; 17: 695-701.
22. Hausser I, Anton-Lamprecht I. Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum Genet 1994; 93: 394-407. [PubMed]
23. Bergqvist D. Ehlers-Danlos type IV syndrome: a review from a vascular surgical point of view. Eur J Surg 1996; 162: 163-170. [PubMed]
24. Watanabe A, Wada T, Tei K, Hata R, Fukushima Y, Shimada T. A novel gene therapy strategy for vascular Ehlers-Danlos syndrome by the combination with RNAi mediated inhibition of a mutant allele and transcriptional activation of a normal allele. Molecular Therapy 2005; 11: 240.
25. Ong K-T, Perdu J, De Backer J, et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet 2010; 376: 1476-1484. [PubMed]
© 2011 Dermatology Online Journal