Familial multiple lipomatosis
Published Web Location
https://doi.org/10.5070/D37088d8pkMain Content
Familial multiple lipomatosis
Brian R Toy MD
Dermatology Online Journal 9(4): 9
From the Ronald O. Perlman Department of Dermatology, New York University
Abstract
Familial multiple lipomatosis is a rare hereditary syndrome with a proposed autosomal-dominant inheritance. A case of an 89-year-old man with this disease is presented, along with his pedigree. Clinical features, genetic evidence, and treatment options are reviewed.
Clinical summary
History.—An 89-year-old man presented to the Dermatology Service of the Department of Veterans Affairs New York Harbor Health Care System for a total body skin examination. Approximately 70 years ago, the patient noted the development of multiple nontender nodules on the trunk and upper extremities. These lesions were slow-growing and became more numerous with age. In fact, he was nicknamed "Lumpy" while serving in the United States Navy during World War II.
Past medical history includes hyperlipidemia, coronary artery disease, and blindness in the left eye secondary to temporal arteritis. Medications include simvastatin, lisinopril, isosorbide dinitrate, and aspirin. Family history includes a subcutaneous disorder in multiple relatives (refer to pedigree).
Physical examination.—Multiple, rubbery, nontender nodules were located on the neck, abdomen, back, and arms. The lesions were mobile, nonfluctuant, and ranged in size from 1-6 cm.
Laboratory data.—Total cholesterol was 202 mg/dl, LDL 133 mg/dl, HDL 50 mg/dl, and triglycerides 97 mg/dl.
Diagnosis.—Familial multiple lipomatosis.
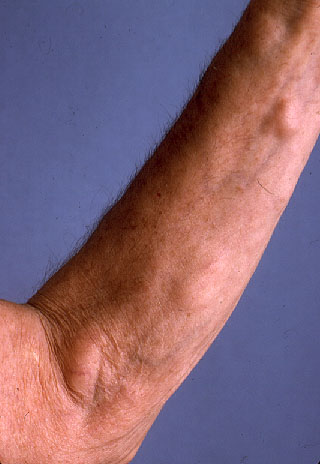
|
| Figure 1 |
|---|
Comment
Familial multiple lipomatosis is a hereditary syndrome of uncertain prevalence. Multiple discrete, encapsulated lipomas are found on the trunk and extremities, with relative sparing of the head and shoulders. The number of tumors in any one patient may vary considerably. Although tumors usually range in size from a few millimeters to 6 cm in diameter, lipomas as large as 25 cm have been reported. [1]. Spontaneous regression and malignant degeneration are rare. The disease is not associated with any abnormalities in lipid metabolism [2].
From case reports, most authors have inferred an autosomal dominant mode of inheritance, although men are affected twice as commonly as women [1]. However, many of these reports are incomplete, as oftentimes the disease does not manifest until after age 30.
Although multiple lipomatosis was first reported in 1846 by Brodie, familial multiple lipomatosis was not reported until 1891 when Blashko described a man, his brother, and several of his children with a similar condition. In 1993 the genetic defect was isolated to chromosome 12q15, which encodes the high-mobility-group protein isoform I-C (HMGIC) [3]. It is now believed that this disorder results from a translocation involving HMGIC on chromosome 12 and the lipoma preferred partner gene (LLP) on chromosome 3 [4]. Although this condition is benign, many patients concerned with cosmesis seek removal of individual tumors. Treatment can include simple excision, endoscopic removal, or liposuction [5].
References
1. Lefell D, Braverman I. Familial multiple lipomatosis. J Am Acad Dermatol 1986;15:275.2. Rubinstein A, et al. Non-symmetric subcutaneous lipomatosis associated with familial combined hyperlipidaemia. Br J Dermatol 1989;120:689.
3. Schoenmakers E, et al. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nature Genet 1995;10:436.
4. Mrozek K, et al. Chromosome 12 breakpoints are cytogenetically different in benign and malignant lipogenic tumors. Cancer Res 1993;53:1670.
5. Ronan S, Broderick T. Minimally invasive approach to familial lipomatosis. Plast Reconstr Surg 2000;106:878.
© 2003 Dermatology Online Journal