Muir-Torre Syndrome / Turcot Syndrome overlap? A patient with sebaceous carcinoma, colon cancer, and a malignant astrocytoma
Published Web Location
https://doi.org/10.5070/D304p4s7fmMain Content
Muir-Torre Syndrome / Turcot Syndrome overlap? A patient with sebaceous carcinoma, colon cancer, and a malignant astrocytoma
Rebecca Kleinerman1 MD, John Marino2 MD, Emmanuel Loucas3 MD
Dermatology Online Journal 18 (5): 3
1. UC Davis Department of Dermatology, Sacramento, California2. New York University, New York, New York
3. Mount Sinai Hospital, New York, New York
Abstract
The Muir-Torre Syndrome is characterized by the clinical constellation of sebaceous neoplasms, keratoacanthomas, and internal malignancies caused by a defect in DNA mismatch repair. Another mismatch repair defect causes Turcot syndrome, which manifests with colorectal and central nervous system neoplasms. We wish to report a case in which the manifestations of both syndromes were observed in the same patient. We further discuss the possible genetic basis for this overlap.
Introduction
The Muir-Torre Syndrome (MTS) represents a predisposition to sebaceous neoplasms, keratoacanthomas, and internal malignancies, most commonly adenocarcinoma of the colon and carcinomas of the genitourinary system. It is an autosomal dominant genodermatosis with variable penetrance that has been related to germline mutations in the MSH2 and MLH1 genes found on chromosomes 3p and 2p, respectively. Both gene products function in DNA mismatch repair. In this correspondence, we describe a patient with MTS by genetic analysis who subsequently developed a cerebral astrocytoma. We use this case to point out that patients with MSH2 or MLH1 mutations and clinical features of MTS may in fact overlap with a closely related condition, Turcot syndrome. Turcot syndrome type I refers to a combination of colorectal adenomas without polyposis and central nervous system neoplasms. Herein, we review the characteristics of both MTS and Turcot syndrome type I in the context of our patient’s presentation, pointing out the genetic evidence for the clinical overlap between the two conditions.
Case presentation
Our patient was a 57-year-old man followed in the practice of the senior author (EL) for skin cancer screening and treatment since 2005. On presentation, he reported a past medical history of stage II colon cancer initially diagnosed in 1997 (at age 46) and treated surgically with resection/omentectomy as well as adjuvant chemotherapy and radiation therapy. His colon cancer recurred in 1999 and he was treated with a sigmoidectomy and chemotherapy. At this time he underwent hereditary non-polyposis colon cancer (HNPCC) testing and was determined to have a MSH2 mutation. In 2001 he was diagnosed with sebaceous carcinoma of the right groin, which was excised without complications. The patient was given a diagnosis of MTS.
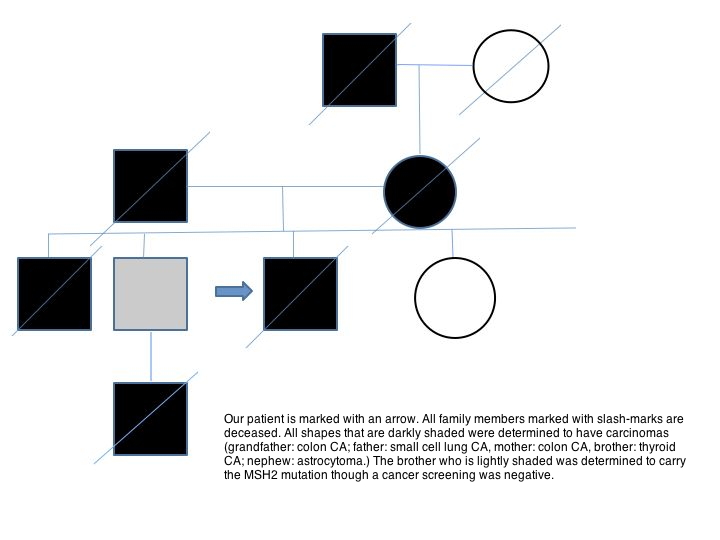 |
| Figure 1 |
|---|
| Figure 1. Genetic tree |
The patient reported that his maternal grandfather died of colon cancer. His father, who was of Polish ancestry, died of lung cancer in his 60s (he was a smoker) and complications of polycythemia vera. His mother, of Scottish/English ancestry also died of colon cancer. One of his brothers died of a cerebral aneurysm at the age of 39 and was found to have thyroid cancer on autopsy. Another brother tested positive for a MSH2 deletion (exons 1-8) at age 60, but an age appropriate cancer screening was negative. A nephew died from complications of a glioblastoma multiforme at the age of 25 (Figure 1).
The patient was diagnosed with a malignant astrocytoma (Grade III) of the frontal lobe in January of 2008, twelve years after his initial diagnosis of colon cancer. He presented with a seizure and was treated with surgery/radiation and adjuvant chemotherapy. He later developed a nodular basal cell of the right nose and the left cheek, treated successfully with Mohs surgery, as well as sebaceous hyperplasia in skin of the of the right ear. In late 2008 the patient once again experienced a recurrence of colon cancer. During the time that chemotherapy was being considered, the patient was found to have a new cerebellar mass consistent with recurrent glioma with leptomeningeal disease. He died soon after from progressive disease.
Discussion
MTS and Turcot syndrome type I are typically described as distinct clinical conditions under the umbrella term of hereditary non-polyposis colorectal cancer syndrome (HNPCC), also known as Lynch syndrome. HNPCC is characterized by an inherited mutation in one of four DNA mismatch repair genes, (MLH1, MSH2, MSH6, and PMS2), predisposing patients to the development of colon cancer and, more rarely, extracolonic carcinoma. The diagnosis of HNPCC is based on a combination of clinical and genetic criteria. Mutations in MLH1 and MSH2 account for the majority of HNPCC cases [1, 2]. MSH2 mutations have been linked to a higher risk for carcinomatosis, especially extraintestinal carcinoma, than MLH1 mutations [3, 4]. Whereas patients with MTS have been shown to have mutations in both the MLH1 and MSH2 genes, patients with Turcot syndrome type I have been described carrying mutations in the MLH1, MSH2 and the PMS2 genes, but primarily are said to have mutations in MLH1 and PMS2 [5].
The gene mutations in HNPCC and, by extension, MTS and Turcot syndrome type I, create an environment of genetic instability in small repeated gene sequences. These genes typically serve in a DNA surveillance capacity, ensuring that base mismatches and insertion-deletion loops are repaired [6]. When an individual inherits a germline mutation in one allele, a second somatic mutation then damages the wild type allele and destroys its tumor suppressor capacity, allowing for the initiation of cancerous change. A microsatellite instability assay (MSI test) is available as a relatively inexpensive screening for the mismatch repair gene mutations associated with HNPCC and related conditions.
The vast majority of patients with MTS have been reported to have mutations in the MSH2 gene [7]. Our patient, with a MSH2 mutation, manifested clinical characteristics of both the MTS and Turcot syndrome type I. Although there is considerable overlap in the gene mutations in both conditions, to our knowledge, there have not been reports of overlap in the clinical constellation of features in one individual. Our patient developed the phenotypic signs of both disorders. We hypothesize that this variation in clinical picture may be related to the specific mutation, e.g., varied splice sites vs. deletions vs. other, though this has not been proven. Some have further suggested that the clinical variability of patients with HNPCC may be related to the distinct expression of modifier genes, both intra and extra-genetic, as well as environmental factors [4]. It should be mentioned that recent advances in the study of gliomas have clarified the molecular pathways that lead to the disease. Whereas our patient passed away prior to the implementation of tests that could clarify the molecular pathogenesis, these may have shed light on the process at hand [8, 9]. Immunohistochemistry searching for an isocitrate dehydrogenase-1 mutation, a gene involved in energy metabolism, is seen in a large number of cases of secondary glioblastomas, and the detection of this mutation has positive prognostic significance [10, 11]. Of note, a recent examination of two gliomas in patients with Turcot syndrome type 1 did not reveal an IDH-1 mutation [10]. Despite the lack of this recent immunohistochemical evidence in the case at hand, our patient’s presentation represents an unusual grouping of clinical manifestations of which the dermatologist following MTS patients should be aware.
References
1. Hampel H, and Peltomaki P. Hereditary colorectal cancer: risk assessment and management. Clin Genet, 2000. 58(2): p. 89-97. [PubMed].2. Mangold E, Pagenstecher C, Leister M, Mathiak M, Rütten A, Friedl W, Propping P, Ruzicka T, Kruse R. A genotype-phenotype correlation in HNPCC: strong predominance of msh2 mutations in 41 patients with Muir-Torre syndrome. J Med Genet, 2004. 41(7): p. 567-72. [PubMed].
3. Vasen HF, Stormorken A, Menko FH, Nagengast FM, Kleibeuker JH, Griffioen G, Taal BG, Moller P, Wijnen JT. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol, 2001. 19(20): p. 4074-80. [PubMed].
4. Lucci-Cordisco E, Zito I, Gensini F, Genuardi M. Hereditary nonpolyposis colorectal cancer and related conditions. Am J Med Genet A, 2003. 122A(4): p. 325-34. [PubMed].
5. Paraf F, Jothy S, Van Meir EG. Brain tumor-polyposis syndrome: two genetic diseases? J Clin Oncol, 1997. 15(7): p. 2744-58. [PubMed].
6. Lee DA, Grossman ME, Schneiderman P, Celebi JT. Genetics of skin appendage neoplasms and related syndromes. J Med Genet, 2005. 42(11): p. 811-9. [PubMed].
7. Ponti G, and Ponz de Leon M. Muir-Torre syndrome. Lancet Oncol, 2005. 6(12): p.980-7. [PubMed].
8. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science 2008. 321: p. 1807-12. [PubMed].
9. Bleeker FE, Molenaar RJ, Leenstra S. Recent advances in the molecular understanding of glioblastoma. J Neurooncol, 2012. [Epub ahead of print]. [PubMed].
10. Lusis EA, Travers S, Jost SC, Perry A. Glioblastomas with giant cell and sarcomatous features in patients with Turcot syndrome type 1: a clinicopathological study of 3 cases. Neurosurgery, 2010. 67(3): p. 811-7; discussion 817. [PubMed].
11. Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, Westphal M, Schackert G, Meyermann R, Pietsch T, Reifenberger G, Weller M, Loeffler M, von Deimling A. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol, 2010. 120(6): p. 707-18. [PubMed].
© 2012 Dermatology Online Journal