Incontinentia pigmenti
Published Web Location
https://doi.org/10.5070/D36vq7m9zrMain Content
Incontinentia pigmenti
Ali Jabbari MD PhD, Jonathan Ralston MD, Julie V Schaffer MD
Dermatology Online Journal 16 (11): 9
Department of Dermatology, New York University, New York, New YorkAbstract
Incontinentia pigmenti is an X-linked dominant genodermatosis that can affect the teeth, eyes, and central nervous system as well as the skin. We describe an infant girl with characteristic cutaneous findings, which progressed through the vesicular, verrucous, and hyperpigmented stages in the first year of life. During the neonatal period, recognition of the linear distribution of vesicular lesions and associated peripheral eosinophilia as well as leukocytosis (which might suggest an infectious etiology) can help to establish the diagnosis. This enables early initiation of ophthalmologic care, which can help to prevent visual sequelae.
History
 |  |
| Figure 1 | Figure 2 |
|---|
 |  |
| Figure 3 | Figure 4 |
|---|
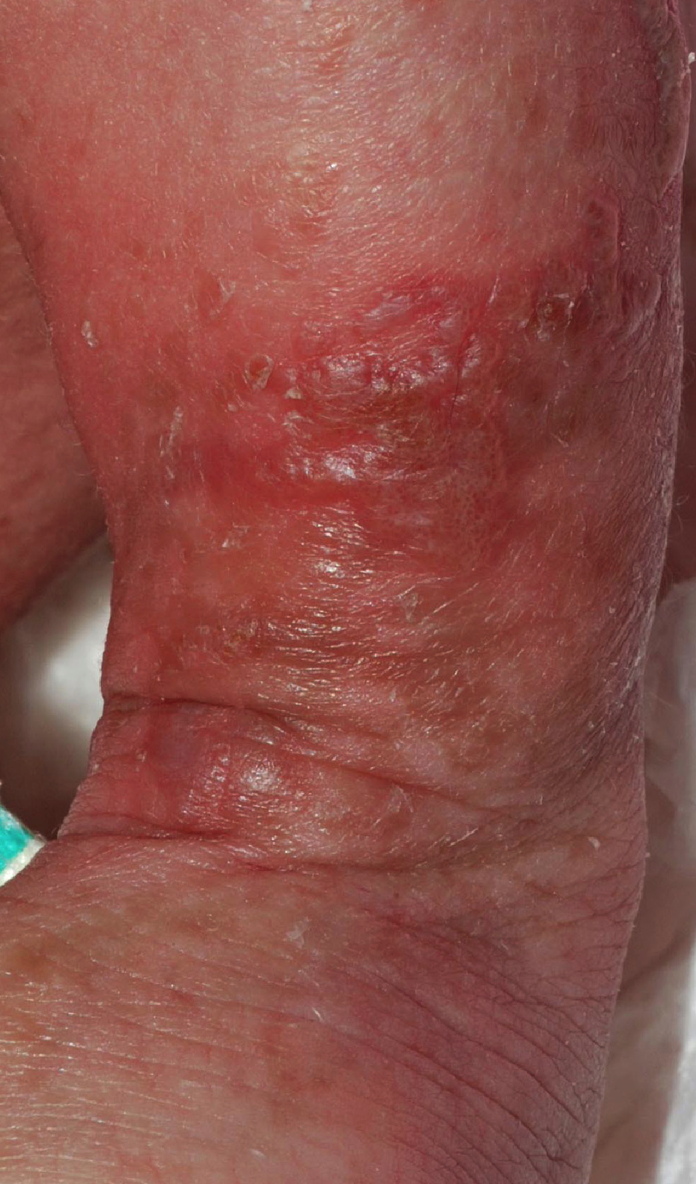
This 15-month-old girl initially presented to the Tisch Hospital Emergency Department at age three weeks for evaluation of widespread blisters that had begun during the first week of life. In the second week of life, she had been hospitalized because of this eruption and associated peripheral blood leukocytosis, but her condition had failed to respond to intravenous antibiotic therapy and cultures (from the blood and skin) were negative. At the Emergency Department, examination by the Pediatric Dermatology Service disclosed linear streaks of erythema with superimposed vesicles on the extremities.
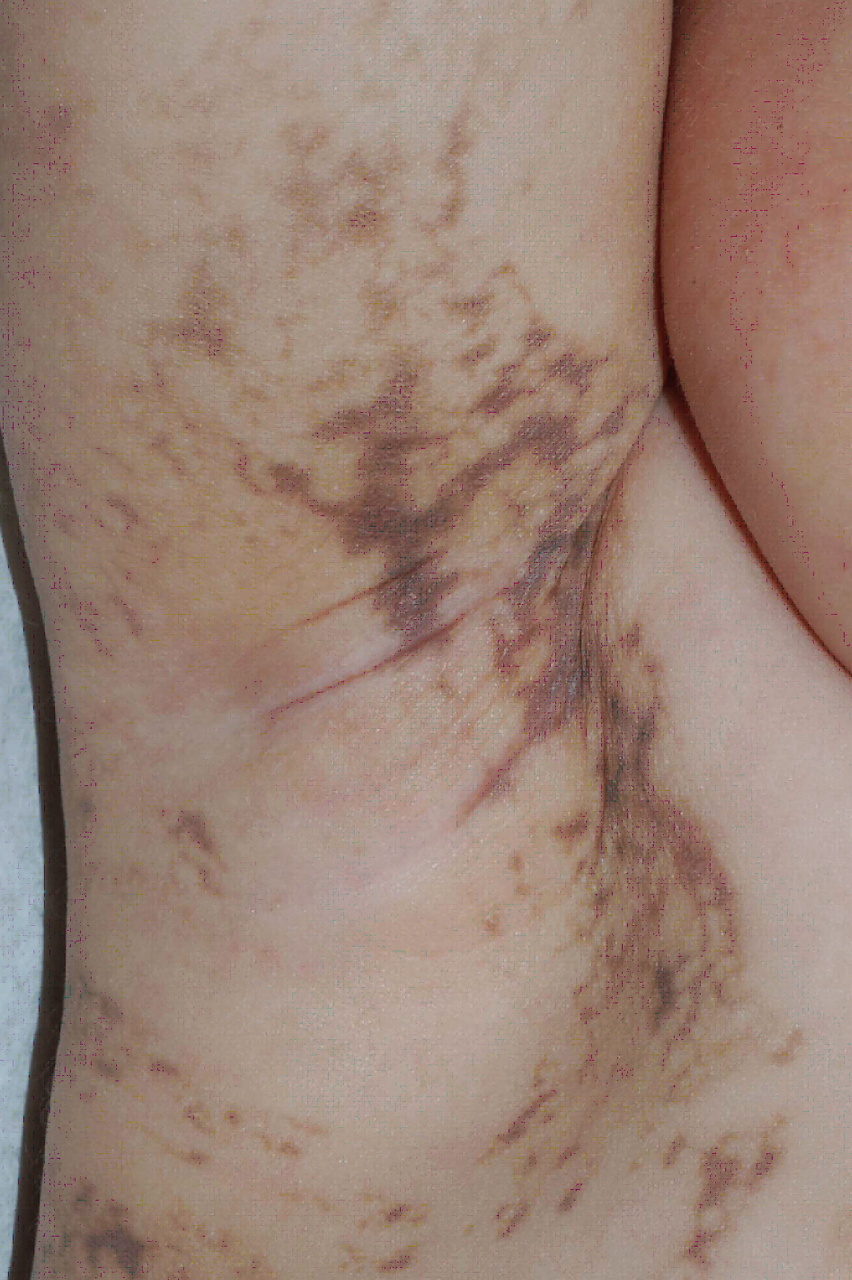
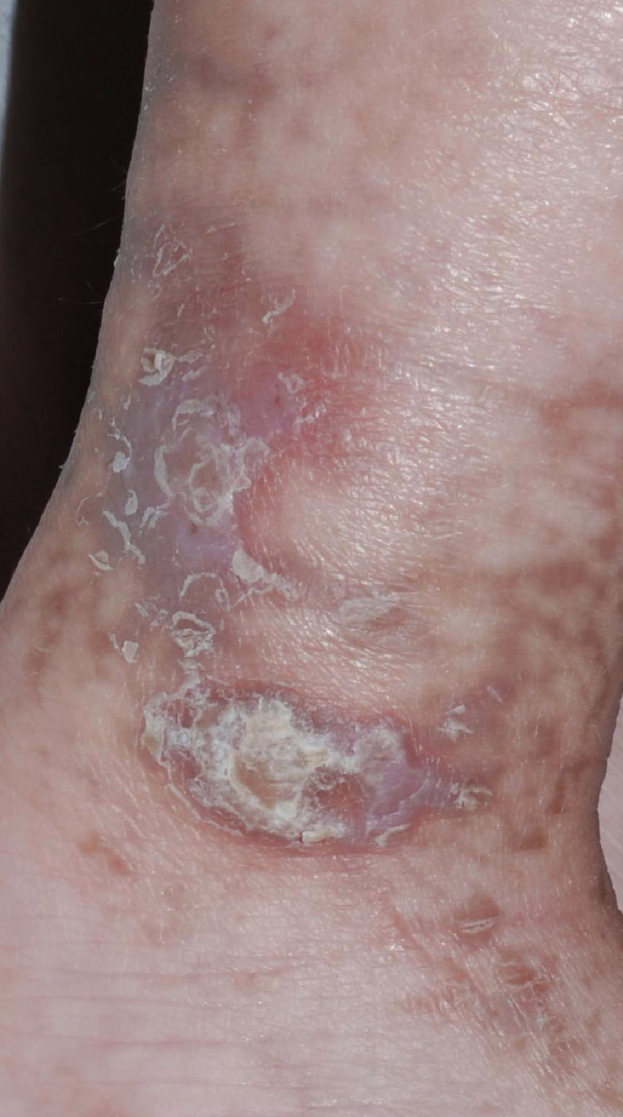
Over the next several months, blisters ceased, and keratotic papules developed in the same distribution. These lesions subsequently faded, and irregular brown streaks progressively appeared on the trunk as well as on the extremities. The patient has had no neurologic problems, and her growth and development have been normal. Serial ophthalmologic examinations have disclosed no abnormalities. There was no family history of skin conditions or dental abnormalities.
Physical examination
Streaks and swirls of gray-brown hyperpigmentation with scalloped borders and a reticulated appearance followed Blaschko lines on the chest, back, groin, axillae, arms, and legs. A few, skin-colored, verrucous papules were present on the ankles and feet. Subtle swirls of relative alopecia were apparent on the posterior scalp. The nails and teeth (two lower incisors) were normal.
Laboratory data
At age three weeks, the white cell count was 23,000/mm³ with 15 percent eosinophils.
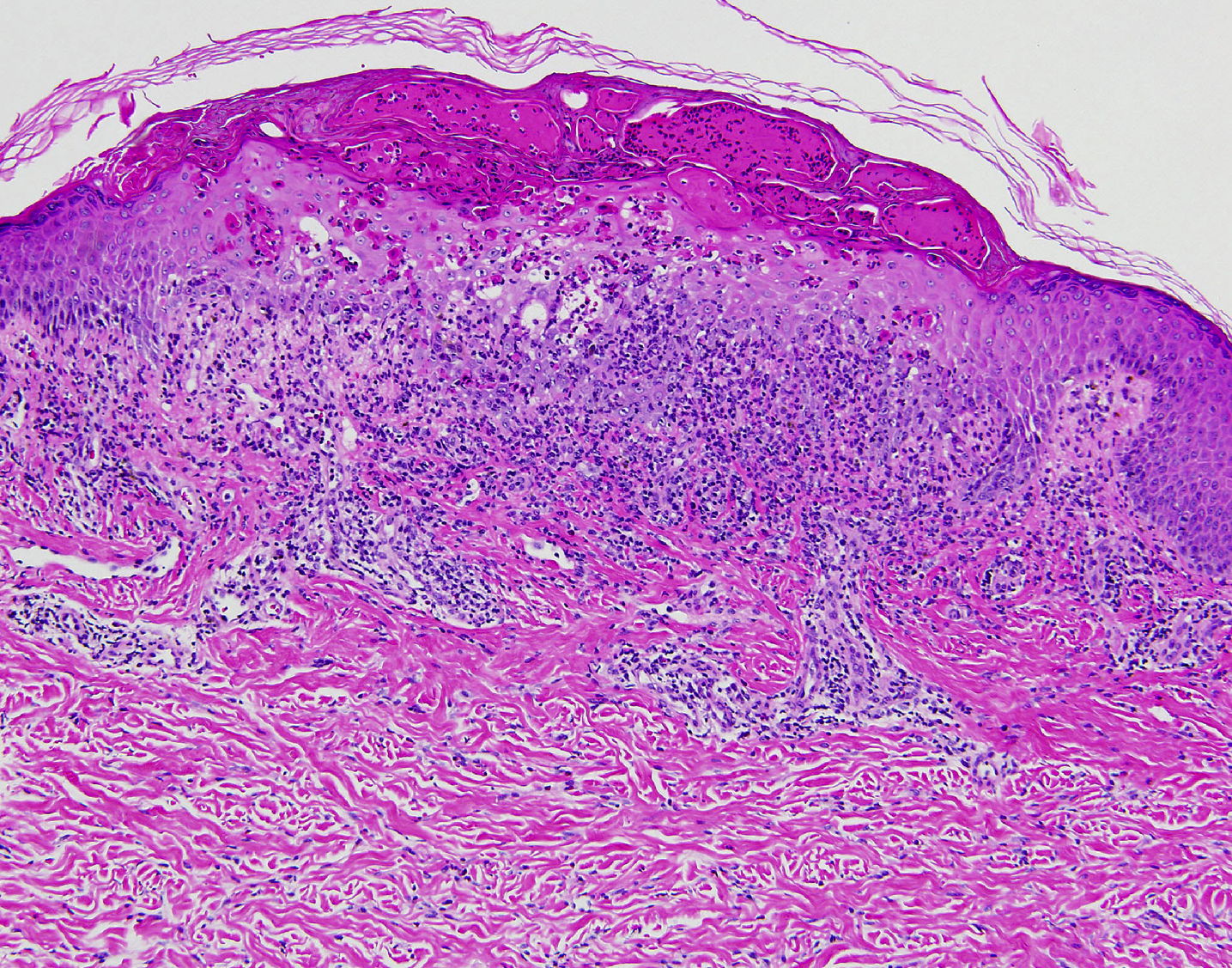
Histopathology
There is a superficial, mid-perivascular, and focal band-like infiltrate of lymphocytes and eosinophils. Some eosinophils and lymphocytes extend into the overlying epidermis where there is vacuolar alteration of the basal layer, slight spongiosis, focal vesicle formation, many individually necrotic keratinocytes, and scale crust. Melanophages are present in the papillary dermis.
Comment
Incontinentia pigmenti (IP) is an X-linked dominant genodermatosis that can affect the teeth, eyes, and central nervous system (CNS) as well as the skin [1]. IP is caused by mutations in the NEMO (NF-κB essential modulator) gene, the protein product of which protects against tumor necrosis factor-α-induced apoptosis [2]. The vast majority of individuals with IP are female patients, since the deletion of NEMO exons 4 - 10 that underlies most cases is lethal prenatally in the hemizygous (XY) state. However, IP can occur in male patients in the setting of mosaicism due to a postzygotic (or half-chromatid) mutation or in association with Klinefelter syndrome (XXY) [3]. Functional mosaicism due to lyonization (random, stably inherited inactivation of one X chromosome in each cell during early embryonic development) explains the distribution of skin lesions along Blaschko lines in girls with IP. The propensity for apoptosis in cells expressing the X chromosome with a NEMO mutation accounts for both the paucity of these cells in the peripheral blood and the resolution of each stage of IP as the abnormal cells are selectively eliminated and replaced by those expressing the normal X chromosome [1, 4].
Cutaneous findings are the most common manifestation of IP and usually represent the presenting signs. They are divided into four overlapping stages: (1) vesiculobullous, which favors the extremities during the first few months of life (but occasionally recurs during childhood in association with a febrile illness); (2) verrucous, which favors the distal extremities in patients one to six months of age (and sometimes adolescents); (3) hyperpigmented, which favors the trunk and intertriginous sites from three months of age through adolescence; and (4) hypopigmented/atrophic, which affects the calves in adolescents and adults [1, 5]. Stage 1 often features peripheral blood leukocytosis and eosinophilia as well as lesional eosinphilic spongiosis, and necrotic keratinocytes are prominent in stages 1 and 2. IP was named for the incontinent pigment that characterizes the gray-brown streaks of stage 3. Scarring alopecia (often swirled at the vertex) and nail dystrophy also can occur.
The extracutaneous manifestations of IP, which are less frequent than are the skin findings, are more likely to cause morbidity [1, 5]. Approximately one-third of patients have ocular involvement, which is characterized by retinal vascular abnormalities that can lead to visual loss during the first year of life [1, 5, 6]. Infants with IP should be referred to a pediatric ophthalmologist for evaluation and close follow-up. CNS abnormalities also occur in one-third of IP patients and can include seizures, developmental delay, and spastic paresis. Neurodevelopmental status should be monitored, and a pediatric neurologist consulted if problems arise. Lastly, dental anomalies, such as delayed eruption, hypodontia and conical teeth affect 50-75 percent of patients, and early dental intervention can help to optimize outcomes [1, 5].
References
1. Berlin AL, et al. Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatol 2002; 47: 169 [PubMed]2. Smahi A, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 2000; 405: 466 [PubMed]
3. Fusco F, et al. Clinical diagnosis of incontinentia pigmenti in a cohort of male patients. J Am Acad Dermatol 2007; 56: 264 [PubMed]
4. Harris A, et al. X inactivation as a mechanism of selection against lethal alleles: further investigation of incontinentia pigmenti and X-linked lymphoproliferative disease. J Med Genet 1992; 29: 608 [PubMed]
5. Hadj-Rabia S, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol 2003; 139: 1163 [PubMed]
6. Goldberg MF. The skin is not the predominant problem in incontinentia pigmenti. Arch Dermatol 2004; 140: 748 [PubMed]
© 2010 Dermatology Online Journal