A rare multisystem disorder: Goltz syndrome – Case report and brief overview
Published Web Location
https://doi.org/10.5070/D373v5d63sMain Content
A rare multisystem disorder: Goltz syndrome – Case report and brief overview
Arpita Jain MD1, Ram Chander MD1, Taru Garg MD1, Nikita MD1, Gurucharan S Shetty MD2
Dermatology Online Journal 16 (6): 2
1. Department of Dermatology, Lady Hardinge Medical College, New Delhi, India. arpitajain_13@yahoo.co.in2. Department of Radiology, Lady Hardinge Medical College, New Delhi, India
Abstract
Focal dermal hypoplasia, also known popularly as Goltz syndrome, is a multisystem disorder characterized by linear or reticulate atrophic macules with fat herniation that is associated with various cutaneous and extracutaneous anomalies. We present a patient with Goltz syndrome who exhibited a classic presentation associated with exophthalmos major and malrotation of the gut. A brief overview of the syndrome is also presented in an attempt to incorporate all associated anomalies reported so far.
Introduction
Goltz syndrome, also known as focal dermal hypoplasia (FDH), is a rare genodermatosis characterized by widespread meso-ectodermal dysplasia. This syndrome was first described in totality by Goltz et al. [1] in 1962, although cases with similar findings had been reported previously as atrophoderma linearis maculosa et papillomatosis congenitalis [2]. It is an X-linked disorder, characterized by the classical cutaneous findings of linear or reticulate atrophic macules, fat herniation, and raspberry papillomas associated with multiple skeletal, ocular, and dental defects.
We report a case of Goltz syndrome with all the characteristic features. Herein we review this syndrome briefly and share our experience on a similar case.
Case report
 |
| Figure 1 |
|---|
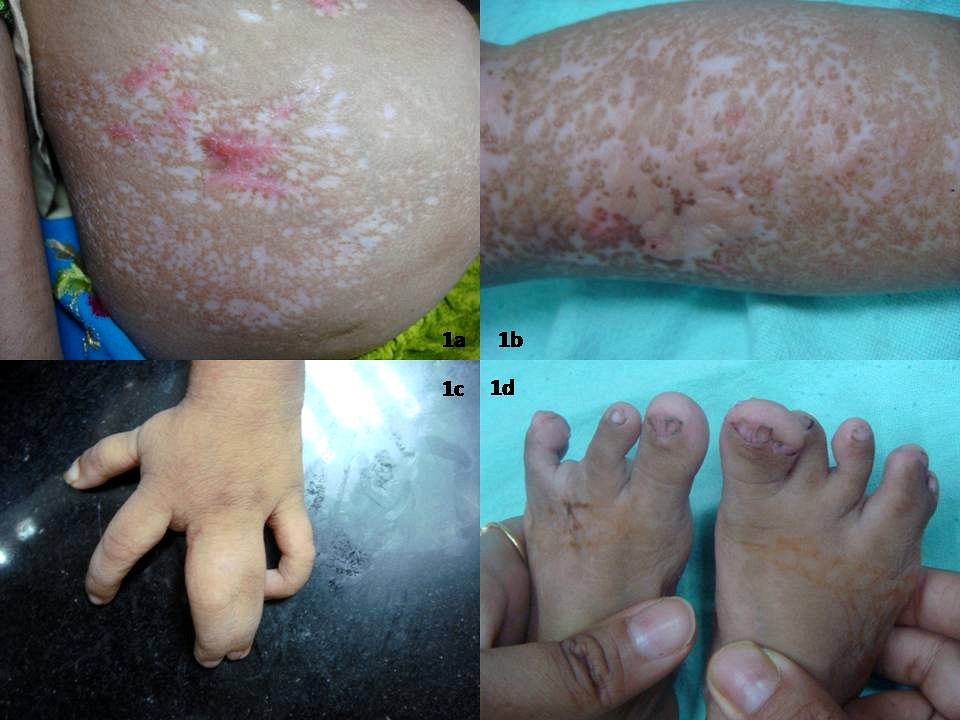
| Figure 1a. Linear and reticulate atrophic plaques with erosions at places Figure 1b. Yellow boggy swellings suggestive of fat herniations through atrophic plaques Figures 1c and 1d. Skeletal deformities of hands and feet |
A 2-year-old girl, the product of a consanguineous marriage, presented to us with mildly pruritic, vermiculate atrophic lesions, in a linear and reticulate pattern over the trunk and extremities. They appeared as linear erosions at birth and gradually healed leaving behind depigmention interspersed with hyperpigmentation (Figure 1a). In addition she had multiple soft boggy yellow to faintly erythematous nodular swellings suggestive of fat herniations, scattered over the extremities, chiefly near the flexors (Figure 1b). Besides this, she had a few small skin colored papillomas over the tips of her digits and a single papilloma over her right tonsillar fossa.
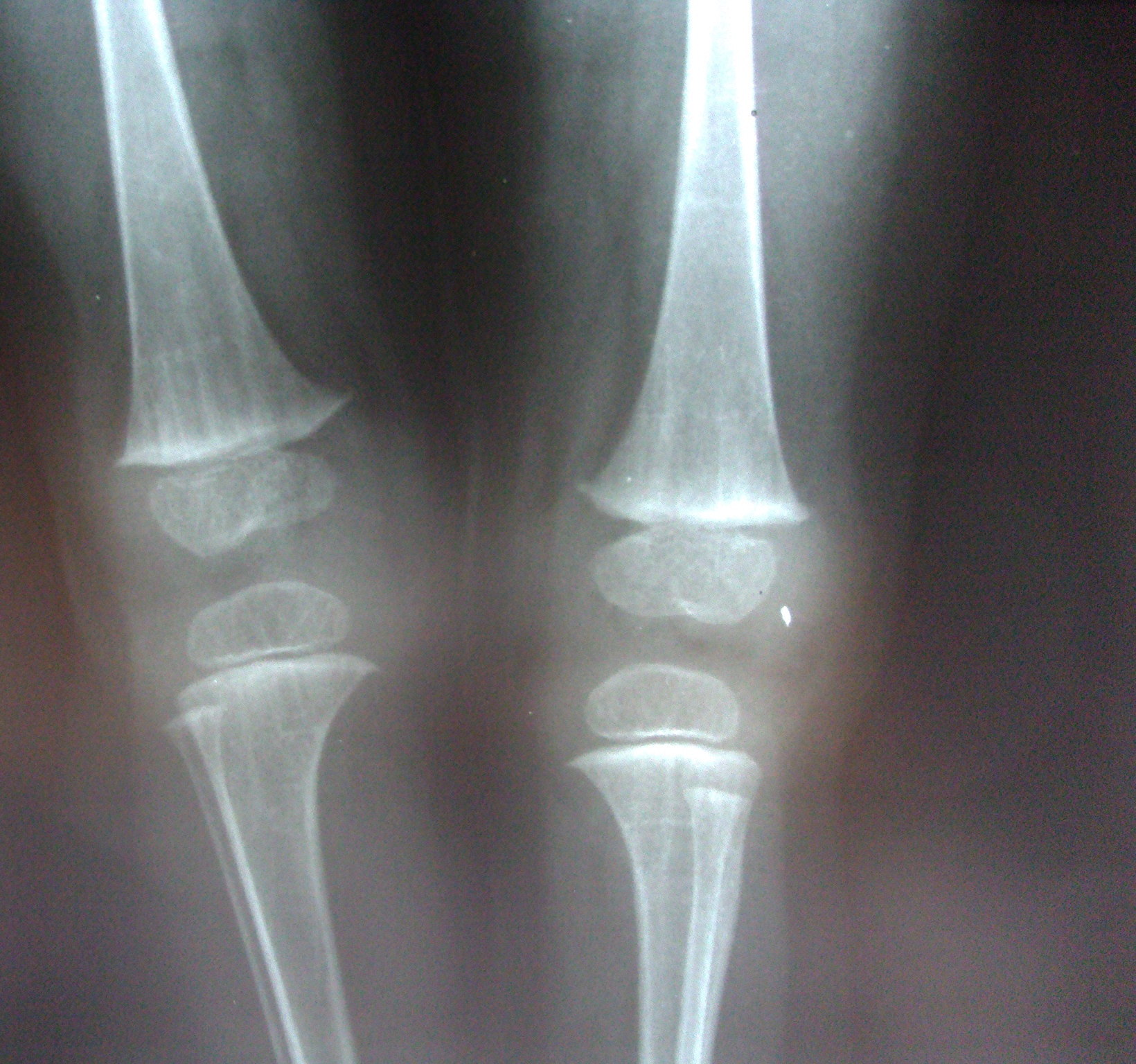
The child had a slim build and short stature. She had typical facies with microcephaly, broad nasal bridge, irregular vermillion border of lips, pointed chin, and large thin, low set ears. She had delayed eruption of teeth, with only four teeth at the age of 2 years. Ophthalmological examination revealed microphthalmia, strabismus, ectropion, and choroidal and iridal colobomas affecting the right eye. The child was born with multiple skeletal defects including syndactyly, polydactyly, and absence of digits (Figures 1c and 1d). A roentegenogram of the long bones showed linear radiodense parallel striations in a juxtra-articular location extending from the metaphysis toward the diaphysis, suggestive of osteopathia striata (Figure 2).
 |  |
| Figure 2 | Figure 3 |
|---|---|
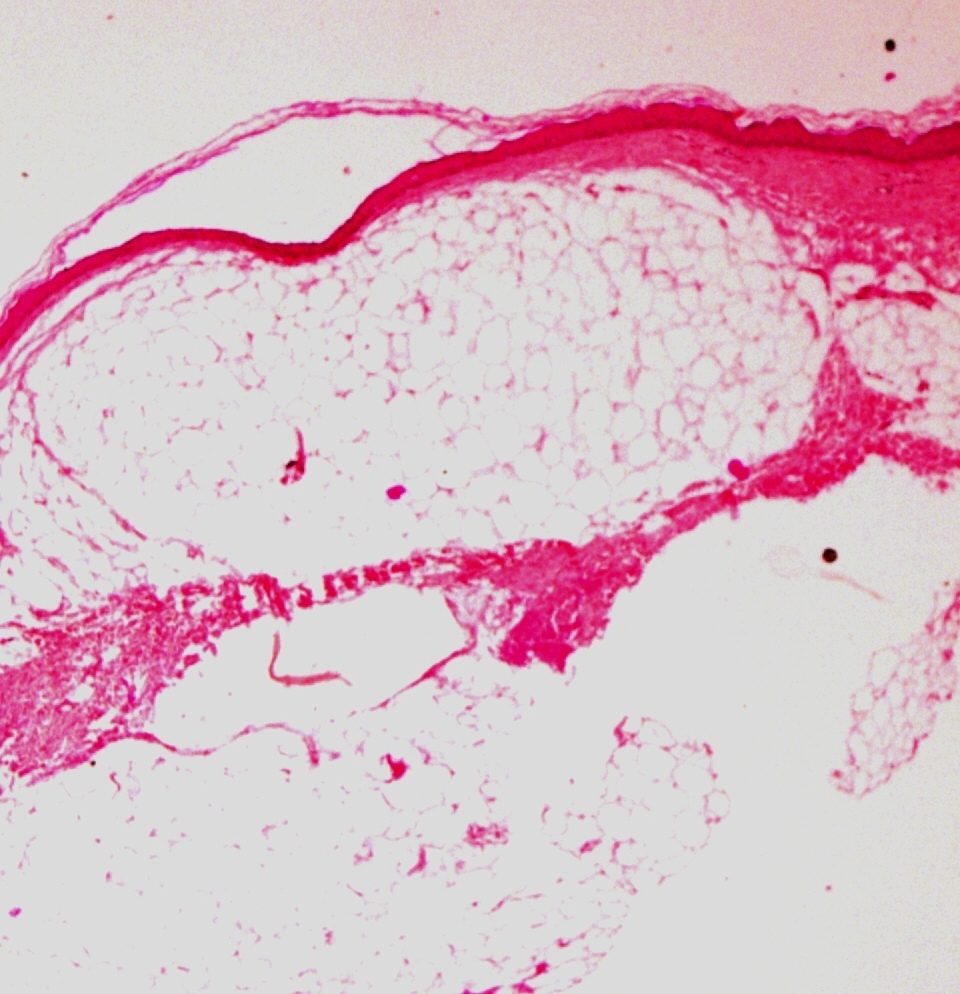
| Figure 2. Linear radiodense parallel striations in a juxtra-articular location suggestive of osteopathia striata Figure 3. Section showing subcutaneous fat separated from epidermis by a thin strand of atrophic dermis (H&E) | |
Associated with the above-mentioned defects, she was born with exophthalmos major and malrotation of the gut, which was surgically corrected. Her developmental milestones were delayed by 6 months. There were no progressive changes in her cutaneous findings. The mother of the child had no history of miscarriages or still births. None of the child’s siblings or family members showed any dermatological, skeletal, or ophthalmological abnormalities.
Histopathology of the atrophic plaque revealed epidermal atrophy with hypoplastic dermis composed of thin collagen bundles. Subcutaneous fat and eccrine appendages were seen just beneath the epidermis, separated only by thin collagen fibers (Figure 3). Based on the clinical presentation and characteristic histopathological, skeletal, and ophthalmological findings, the diagnosis of Goltz syndrome was made.
Discussion
Focal dermal hypoplasia syndrome involves all the three tissue layers, namely ectoderm (skin, eyes), mesoderm (bones, teeth) and endoderm (various mucosas). The syndrome has a variable expression ranging from mild to severe mutilating manifestations.
It is an X-linked dominant disorder with a mosaic pattern, mainly affecting females. In-utero lethality in males is evidenced by frequent reports of miscarriages and still-births and no reports of transmission from father to son. Ten to fifteen percent of cases have been reported to occur in males, though this figure may be an exaggerated one because of higher reporting in males [3]. Those males who survive could be a result of somatic mosaicism or a sporadic new mutation. Such cases have always been reported to be the first affected cases in the family [3]. Mosaicism for mutations in the PORCN gene on chromosome Xp11.23 has recently been implicated as the genetic basis of FDH [4]. Variations in the clinical severity is said to be partly due to lyonization of the X chromosome and partly because of post-zygotic genomic mosaicism; 95 percent of cases are sporadic. Post zygotic somatic mosaicism is thought to be responsible for such sporadic cases [5].
The predominant feature of FDH is connective tissue dysplasia, particularly of the skin and skeletal system. Uitto and coworkers have reported a defect in fibroblast growth characteristics in the affected skin, thereby leading to a dystrophic dermis and focal herniation of subcutaneous fat [6]. Buchner and Itin also showed absence of collagen IV from the dermo-epidermal interface [7]. Histopathology of the atrophic and lipomatous skin reveals marked reduction in the thickness of the dermis. Collagen fibers are attenuated and fat cells extend virtually up to the epidermis; thin strands of dermal connective tissue may be interspersed [5].
Cutaneous lesions are usually regarded as an essential component of FDH, although there are a few reports of the syndrome without any skin lesions [8]. Red, hypopigmented or depigmented atrophic macules, arranged in a linear or blaschkoid pattern or in a reticulate grouping, may be observed anywhere on the body and are often noted at birth. Telengiectasias are commonly seen interspersed between atrophic plaques. Skin abnormalities may either be localized or may be extensive. At birth the lesions may present as blisters or erosions, which soon heal to leave behind typical atrophic scars. At times there is total absence of skin that relates to a profound defect in the dermal tissue, which is unable to support the epidermis and appendages [3]. The skin lesions might be pruritic and photosensitive [5]. The second characteristic cutaneous feature is fat herniation, which appears as soft, pink-brown nodules overlying the thin atrophic skin, mostly on the antecubital and popliteal fossae. These may appear any time after birth [7]. Other characteristic cutaneous findings include raspberry papillomas, present mostly at the junction of skin and mucosa, around lips, or the vulvar and perianal areas. Our case showed the characteristic cutaneous features, but had papillomas over her toes and in the oral cavity. A case of unilateral FDH has also been recently reported [9].
A child with FDH often has characteristic facies, like our case, with a triangular face and pointed chin. The nasal bridge is narrow with a broad nasal tip and unilateral notching of the nasal alae. The ears are thin, protruding, and low set. The face may be asymmetric with hemi-hypertrophy. Besides skin, skeletal, ocular, and dental systems are most commonly affected; defects are seen in variable combinations in the majority of cases. The most common findings in these systems are tabulated in Table 1. The other organ systems of the body also may show some deformities. These rare associations are compiled together in Table 2.
Focal dermal hypoplasia is a syndrome with a distinct constellation of multisystem defects. Suspicion for the syndrome must arise whenever a child, especially female, presents with generalized, or at times localized, areas of atrophic plaques in a linear and blashckoid pattern along with soft boggy swellings suggesting fat herniation. These findings are almost always associated with profound and obvious skeletal defects mostly over the hands and feet. Various types of hernias and exophthalmos may be presenting complaints, as seen in our case. All systems should be thoroughly screened to look for any other associated anomalies mentioned in Table 1 and Table 2. Histopathology is the most specific diagnostic technique for diagnosing FDH. Gene sequence analysis for detection of the PORCN gene mutation can also be carried out. It was not performed in our case because the clinical, radiological, and histopathological findings were confirmatory and the parents did not give their consent.
The most common differential diagnoses for FDH are MIDAS (microphthalmia, dermal aplasia, sclerocornea), incontinentia pigmenti, and Rothmund Thomson syndrome. Other conditions to be considered are Adams-Oliver syndrome, nevus lipomatoides superficialis, aplasia cutis and Ecrodactyly Ectodermal Dysplasia-Clefting (EEC) syndrome. MIDAS syndrome shows dermal atrophy typically limited to the upper half of the body without any fat herniation or other defects associated with FDH.
Incontinentia pigmenti can be easily ruled out because of the presence of atrophic lesions and absence of all of the previous stages of the disorder. The presence of atrophic plaques at birth almost rules out Rothmund Thomson syndrome because it generally presents after 3 months of age and the lesions are not linear. The presence of a generalized distribution rules out Adams-Oliver syndrome, which is always localized to the scalp and skull. Nevus lipomatoides has similar histopathological findings to FDH, but exhibits only a localized group of soft fleshy nodules over the lower back without any other cutaneous and extracutaneous features. Aplasia cutis is generally a solitary lesion with absence of other defects seen commonly in FDH. EEC syndrome is differentiated by the presence of lobster claw-type deformities and clefting of the lip and palate. Absence of intrauterine infections should be ascertained to rule out congenital infections as a cause of cutaneous defects.
Focal dermal hypoplasia is a multisystem disorder, which requires multi- specialty care. The child usually leads a normal life with near normal intelligence. Flashlamp-pumped pulsed dye laser may be useful in alleviation of pruritus and partial response in skin lesions. Papillomas can be removed by cryotherapy or cauterization, but may recur again. Proper genetic counselling, emphasising the X-linked dominant inheritance, should be provided for the parents of affected offspring. Prenatal diagnosis is not possible, but ultrasonography can pick up some of the radiological abnormalities. The carriers can be traced by genetic mapping. Osteopathia striata may be present in 20 percent of gene carriers and they may show only non-cutaneous defects associated with Goltz syndrome.
References
1. Goltz RW, Peterson WC, Gorlin RJ et al. Focal dermal hypoplasia. Arch Dermatol. 1962; 86: 708-17. [PubMed]2. Liebermann S. Atrophoderma linearis maculosa et papillomatosis congenitalis. Acta Dermatol Venereol. 1935; 16: 476-84.
3. Goltz RW. Focal dermal hypoplasia syndrome: an update. Arch Dermatol. 1992; 128: 1108-111. [PubMed]
4. Leoyklang P, Suphapeetiporn K, Wananukul S, et al. Three novel mutation in the PORCN gene underlying focal dermal hypoplasia. Clin Genet. 2008; 73: 373-79. [PubMed]
5. Temple IK, MacDowell P, Baraister M et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990; 27:180-87. [PubMed]
6. Uitto J, bauer EA, Santa Cruz DJ et al. Focal dermal hypoplasia: abnormal growth characteristics of skin fibroblasts in culture. J Invest Dermatol. 1980; 765: 170-75. [PubMed]
7. Buchner SA, Itin P. Focal dermal hypoplasia syndrome in a male: case report, histologic and immunohistochemical studies. Arch Dermatol. 1992; 128: 1078-82. [PubMed]
8. Ayme S, Fraser FC. Possible examples of the Goltz syndrome (focal dermal hypoplasia) without linear areas of skin hypoplasia. Birth defects. 1982; 18: 59-65. [PubMed]
9. Aoyama M, Sawada H, Shintani Y et al. Case of unilateral focal dermal hypoplasia(Goltz syndrome). J Dermatol. 2008; 35: 33-5. [PubMed]
10. Hall EH, Terezhalmy GT. Focal dermal hypoplasia syndrome. J Am Acad Dermatol. 1983; 9: 443-41. [PubMed]
11. Vakilzadeh F, Happle R, Peters P et al. Focal dermal hypoplasia with apocrine nevi and striations of bones. Arch Dermatol Res. 1976; 256: 189-95. [PubMed]
12. Al Haddad C, Maybodi M, Butrus S et al. Congenital symblephara, progressive corneal pannus and skin defects. J Pediatr Ophthalmol Strabismus. 2009; 46: 168-70. [PubMed]
13. Ascherman JA, Knowles SL, Troutman KC. Extensive facial clefting in a patient with Goltz syndrome: multidisciplinary treatment of a previously unreported association. Cleft Palate craniofac. J 2002; 39: 469-73. [PubMed]
14. Irvine AD, Stewart FJ, Bingham EA, Nevin NC, Boston BE. Focal dermal hypoplasia (Goltz syndrome) associated with intestinal malrotation and medistinal dextroposition. Am J Med Genet. 1996; 62: 213-15. [PubMed]
15. Reddy J, Laufer MR. Congenital anomalies of the female reproductive tract in a patient with Goltz syndrome. J Pediatr adolesc Gynecol. 2009; 22: 71-72. [PubMed]
© 2010 Dermatology Online Journal