Hereditary hemorrhagic telangiectasia
Published Web Location
https://doi.org/10.5070/D342v489fgMain Content
Hereditary hemorrhagic telangiectasia
Nathalie Q Nguyen MD
Dermatology Online Journal 11 (4): 19
Department of Dermatology, New York University School of Medicine
Abstract
A 45-year-old man with hereditary hemorrhagic telangiectasia (HHT) presented with numerous mucocutaneous telangiectases, recurrent nosebleeds, and several first degree relatives with similar symptoms. HHT has been linked to mutations in the genes endoglin and activin receptor-like kinase-1 (ALK-1), which are located on chromosome 9q33-34 and 12q13, respectively. The resultant proteins mediate binding and signaling of transforming growth factor-β. The clinical features, molecular basis, and management of HHT are reviewed.
A 45-year-old man first noticed numerous telangiectases on his lips and tongue as a child. As the patient grew older, the number of telangiectases increased, and he experienced recurrent nosebleeds. He currently suffers from approximately five nosebleeds per month. Several years ago, the patient noticed telangiectases under his fingernails as well as an increased number of telangiectases on his face and chest. His mother, grandmother, uncle, and two brothers also have numerous telangiectases and recurrent nosebleeds. The patient denies a history of gastrointestinal bleeding, pulmonary abnormalities, or cerebral hemorrhages.
On examination he was found to have multiple, 2-3-mm, bright-red macules scattered on the lips and tongue. On the face and upper chest, there were numerous telangiectases. Under the patient's fingernails, several pinpoint erythematous macules were observed.
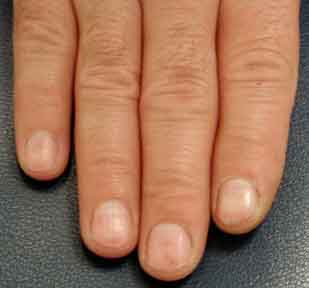
|
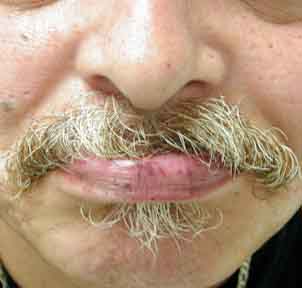
|
| Figure 1 | Figure 2 |
|---|
Hemoglobin was 13.8g/dL, hematocrit 40.9 percent, and mean corpuscular volume 89.3fL. The liver function tests and complete metabolic panel were normal. A colonoscopy and bronchoscopy performed approximately 5 years earlier failed to show any arteriovenous malformations.
Comment
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal dominant vascular disorder that affects approximately 1 in 5,000-8,000 individuals [1]. HHT, first recognized in the nineteenth century, is a disorder of arteriovenous malformations (AVM) and telangiectases. Patients develop frequent episodes of bleeding, increasing with age, especially in the form of recurrent nosebleeds. Nearly all patients will develop mucocutaneous telangiectases. Approximately 30 percent of HHT patients have pulmonary AVMs, 30 percent have hepatic involvement, 10-20 percent have cerebral involvement, and 15 percent will develop gastrointestinal bleeding [2].
HHT is a heterogeneous disease both between families and among members of a single family. Genetic linkage studies have mapped HHT to chromosome 9q33-34 in some families (HHT type 1) and 12q13 in others (HHT type 2) [3]. The mutated genes at these loci have been identified as endoglin and activin receptor-like kinase-1 (ALK-1), respectively. They mediate binding and signaling of the transforming growth factor-β (TGF-β) superfamily of proteins, which includes TGF-βs, activins, and the bone-morphogenetic proteins. ALK-1 is a TGF-β superfamily type I receptor, and endoglin associates with different signalling receptors and can modify TGF-β-1 signalling. The expression of these two proteins appears to be limited to the vascular endothelium. Endothelial cells derived from HHT1 or HHT2 patients express approximately one half of the normal endoglin or ALK-1 levels, respectively. Therefore, it is believed that in most cases, HHT results from endoglin or ALK-1 haploinsufficiency [3].
The Curacao criteria utilizes the clinical features of epistaxis, multiple telangiectases, visceral AVM, and a first degree relative with HHT [4]. A diagnosis of HHT is considered definite if three criteria are present, possible if two criteria are present, and unlikely if fewer than two criteria are present. Management of recurrent nosebleeds includes supplementation with iron for anemia, laser ablation, surgical correction (septal dermatoplasty, septal closure, or arterial ligation), embolization, and prevention with the use of nasal sprays [1]. Gastrointestinal bleeding typically occurs in the fifth or sixth decade and often presents as an iron-deficiency anemia. Laser ablation and surgical correction have been used, but the only therapy supported by evidence from randomized, controlled trials is the use of female hormones in transfusion-dependent patients [5]. Screening methods for pulmonary AVMs include thoracic imaging or detection of right-to-left shunting. Patients with pulmonary AVMs are at risk not only for hemorrhage but also for paradoxical embolism. Screening of asymptomatic HHT patients for cerebral AVMs remains highly controversial.
References
1. Begbie MD, et al. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgrad Med J 2003;79:182. Haitjema T, et al. Screening family members of patients with hereditary hemorrhagic telangiectasia. Am J Med 1995; 99: 519
3. van den Dresche S, et al. Hereditary hemorrhagic telangiectasia: an update on transforming growth factor beta signaling in vasculogenesis and angiogenesis. Cardiovasc Res. 2003;58:20
4. Shovlin CL, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu). Am J Med Genet 2000; 91:66
5. Van Cutsem E, et al. Treatment of bleeding gastrointestinal vascular malformations with oestrogen-progesterone. Lancet 1990;335:935
© 2005 Dermatology Online Journal