Behçet's Disease
Published Web Location
https://doi.org/10.5070/D33hb3h6v3Main Content
Behçet Disease
Fitzgeraldo A Sanchez-Negron MD
Dermatology Online Journal 10 (3): 14
From the Ronald O. Perelman Department of Dermatology, New York University
Abstract
A 29-year-old man presented with oral and genital ulcers, erythematous papules and pustules on his back and chest, and deep vein thrombi. A diagnosis of Behçet disease was made. Behçet disease is a relapsing disorder that affects the mucocutaneous surfaces. It presents usually as ulcers on the orogenital mucosae, but can also present as an acneiform eruption. The International Study Group on Behçet disease has established criteria that consist of oral and genital lesions, ocular involvement, skin findings, and a pathergy. Treatment of choice is colchicine or prednisone.
Clinical synopsis
History.—A 29-year-old man had a 12-year history of oral and penile ulcers. The patient moved from Morocco 1 year ago, and since then has had multiple hospital admissions and visits to the emergency room for these painful lesions. He has noted a red eruption on his chest and back for the past week prior to admission. He had noted these lesions in the past with the ulcers. On admission the patient had experienced joint pain of the knees for the past 3 days with a temperature of 101.9° F. There were no ocular findings.
His past medical history includes deep venous thrombi and Budd-Chiari syndrome. Medications include warfarin, colchicine, and dexamethasone.
Physical Examination.— Erythematous papules and pustules were present on the chest and back. A 1-cm erosion was noted on the tonsil, and a 5-mm erosion was noted on the glans penis. Tender lymphadenopathy was present in the right axilla and right submandibular area.
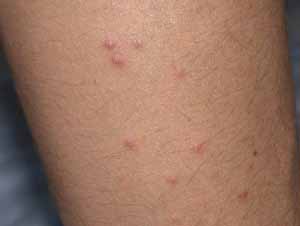
|
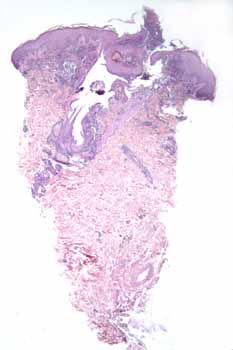
|
| Figure 1 | Figure 2 |
|---|
Laboratory data.— A complete blood count and blood chemistry profile were normal. Blood cultures showed no growth. A chest radiograph was negative. An abdominal computed-tomography scan showed thrombi in the right hepatic vein, inferior vena cava, left renal vein, and iliac veins.
Histopathology.— There is a disrupted folliculosebaceous unit surrounded by neutrophils, histiocytes, and foci of fibrosing granulation tissue. Neutrophils permeate the infundibulum and overlying mounds of parakeratosis. There is leukocytoclasis but no evidence of vasculitis or thrombosis. A periodic-acid-Schiff stain is negative for fungi.
Diagnosis.—Behçet Disease
Comment
In 1936, Behçet described a patient with eye disease and oral and genital ulcers. Mostly seen in patients of Japanese, Middle Eastern, or Mediterranean origin, Behçet disease is thought to have both genetic and environmental influences. The Asian population with the syndrome has an 81 percent chance of having the HLA-B51 allele.
Behçet disease is a predominantly neutrophilic disorder that presents with elevated levels of tumor necrosis factor and IL-8. There is also an increased expression of CD94, which suggests that NK receptors play a pathogenic or regulatory role. [1]
The criteria for the diagnosis of Behçet disease have evolved over the years, and in 1990 the International Study Group on Behçet disease established the following criteria for the diagnosis: [2,3] recurrent oral ulcerations: (aphthous) recurring at least three times in one 12-month period, plus any two of the following: recurrent genital ulceration: aphthous ulcers with scars, observed by subject or physician; eye lesions: anterior or posterior uveitis, cells in the vitreous by slit-lamp examination or retinal vasculitis observed by an ophthalmologist; skin lesions: erythema nodosum-like, papulopustular lesions, or distinct acneiform nodules in postadolescent patients not on glucocorticoids; or pathergy: observed at 24-48 hours by a physician.
Aphthous stomatitis is usually the first presenting sign of the disease, and it may precede other symptoms by many years. Perianal and genital ulcers tend be deeper and more painful. A unique syndrome of both Behçet disease and relapsing polychondritis has been recognized and named MAGIC syndrome (mouth and genital ulcers with inflamed cartilage). [4]
The main cause of morbidity is eye involvement, which occurs in 90 percent of cases. Blindness is the worst complication, but conjunctivitis, scleritis and keratitis are also observed. Vascular disease may occur as aneurysms, deep vein thrombi, cardiac valvular involvement, and myocarditis.
Treatment depends on the area involved. For mucocutaneous areas, colchicine and oral glucocorticoids are used. For severe cases, thalidomide has been tried as well as methotrexate. For systemic disease, prednisone and azathioprine have been successfully used as well as cyclosporine, chlorambucil, IVIg, and mycophenolate mofetil. Infliximab has been used with unimpressive results. [5]
References
1. Saruhan-Direskeneli G, Uyar FA, Cefle A, Onder SC, Eksioglu-Demiralp E, Kamali S, Inanc M, Ocal L, Gul A. Expression of KIR and C-type lectin receptors in Behcet's disease. Rheumatology (Oxford). 2004 Apr;43(4):423-7. Epub 2003 Dec 16. PubMed2. International Study Group for Behçet's Disease. Criteria for diagnosis of Behçet's Disease. Lancet 1990; 335:1078.
3. Tunc R, et al. A reassessment of the International Study Group criteria for the diagnosis (classification) of Behçet's syndrome. Clin Exp Rheumatol 2001; 19 (Suppl 24):545.
4. Imai H, et al. Mouth and genital ulcers with inflamed cartilage (MAGIC syndrome): a case report and literature review. Am J Med Sci 1997; 314:330.
5. Yucel AE, et al. Failure of infliximab treatment and occurrence of erythema nodosum during therapy in two patients with Behçet's disease. Rheumatology 2004; 43:394.
© 2004 Dermatology Online Journal