A 71-year-old man with spindle-cell neoplasm of unknown origin: A difficult-to-diagnose clear-cell sarcoma
Published Web Location
https://doi.org/10.5070/D359f6c9t5Main Content
A 71-year-old man with spindle-cell neoplasm of unknown origin: A difficult-to-diagnose clear-cell sarcoma
Lori Spencer PhD1, and Drazen Jukic MD, PhD1, 2
Dermatology Online Journal 10 (1): 8
1. University of Pittsburgh School of Medicine2. University of Pittsburgh Physicians, Department of Dermatology; University
of Pittsburgh Physicians, Department of Pathology
Abstract
A patient initially presented in 1988 with a solitary axillary mass, diagnosed as a high-grade neuroendocrine spindle-cell neoplasm; there was no history of a primary cutaneous malignancy. After subsequent development of a pulmonary nodule in 2001 (14-years post initial diagnosis), the case was reviewed and the possibility of metastatic melanoma was raised. The histopathologic and immunohistochemical profile of this melanocytic neoplasm was diagnostic of clear cell sarcoma (CCS) of tendons and aponeuroses, although the differential diagnosis included malignant melanoma, follicular dendritic and interdigitating cell tumors, malignant peripheral nerve sheath tumor, and a category of so-called PEComas. It is the role of pathologists, particularly dermatopathologists, to distinguish CCS from malignant melanoma, and to alert the clinician, because proper diagnosis ultimately influences treatment. We discuss the immunophenotype, differential diagnosis, and molecular signatures of these neoplasms, and review the pertinent literature on these entities.
Introduction:
In 1988 a 58-year-old white man noticed a lump in his right axillary region. He underwent an excisional biopsy, which revealed a 3-cm firm nodular lesion. The mass was well circumscribed and was considered to be a lymph node replaced either by metastatic neoplasm or granulomatous disease (Figs. 1 and 2). The working diagnosis was that of a neoplasm of endocrine or neuroendocrine origin. No further therapy was performed at that time.
In 1999 he was found to have an elevated PSA (prostate specific antigen) and an abnormal digital-rectal exam. After a transrectal needle biopsy (Fig. 3), diagnosis of prostate cancer was confirmed and a prostatectomy was performed (Fig. 4).
In 2001, at the age of 71, this patient presented to our clinic with a chief complaint of chest pain. He noted that the chest pain initially began after a car accident. At approximately the same time he started experiencing hematuria. At that time, both a chest x-ray and CT scan were performed, revealing a left lower lung lobe tumor; on bronchoscopy and fine needle aspiration (FNA) this was interpreted as a possible carcinoid tumor (Figs. 5 and 6). However, the results of an OctreoScan argued against metastatic disease of primary neuroendocrine differentiation.
The patient experienced 10-pound weight loss within the 4 weeks. He was also experiencing bone pain all over his body, blood in his stools, and constipation.
As a diagnostic and treatment procedure, he underwent left exploratory thoracotomy, and excisional biopsy of the implant in parietal pleura, and pleurectomy for tumor debulking from the diaphragmatic surface (Figs. 7, 8, and 9).
Later that year the patient was noted to have persistent pleural effusion, which was aspirated yielding 3 liters of serosanguinous fluid. After the procedure, the left lung expanded, but there were numerous areas of atelectasis. His pulmonary function did not improve, and he continued to become more dyspneic with a decreased mental status throughout. He eventually passed away from respiratory failure.
Histopathology
|
|
| Figure 1 | Figure 2 |
|---|---|
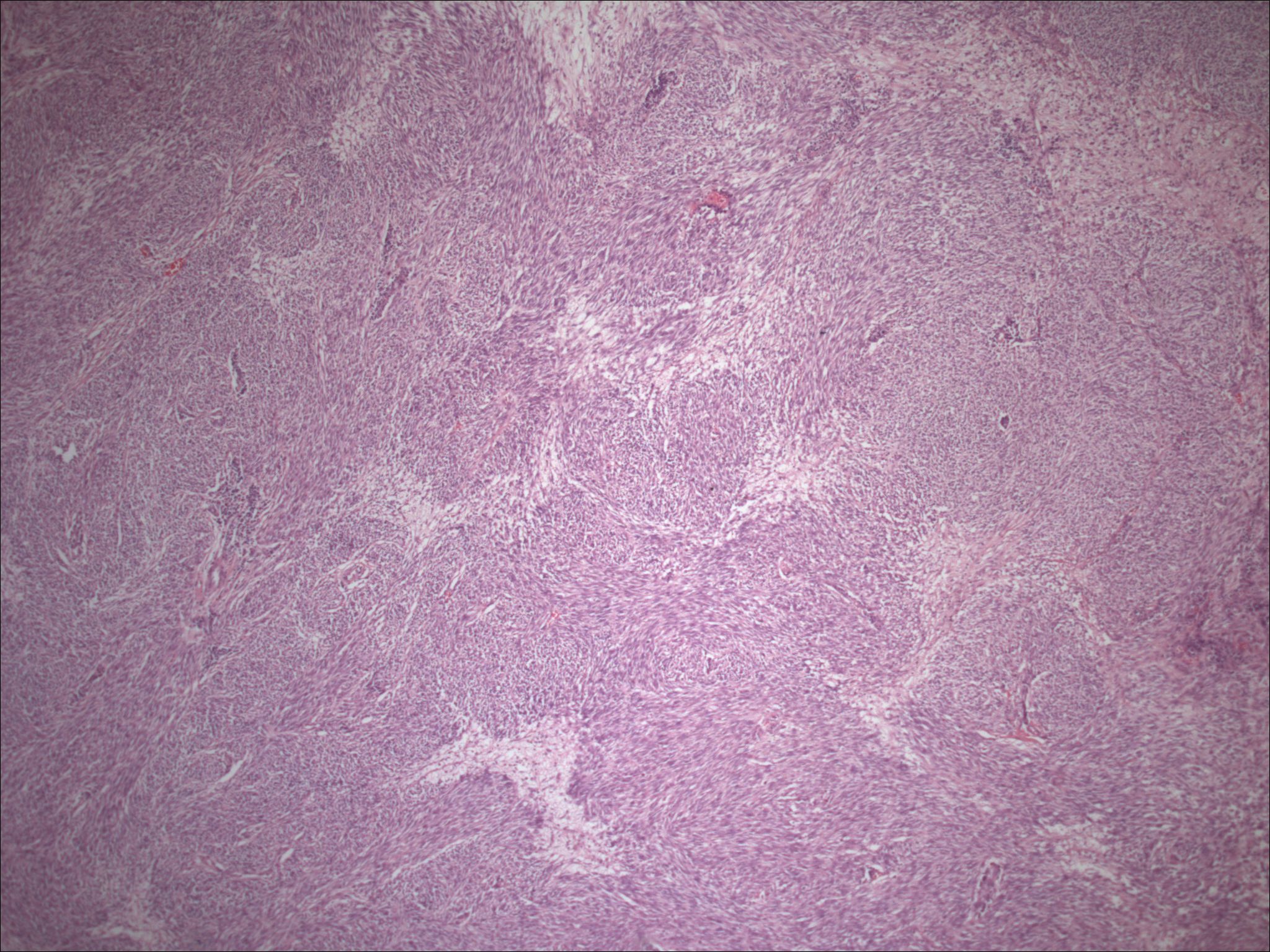
| Figure 1. Low power view of the patient's initial specimen from 1988, which reveals a predominately bland appearing spindle cell neoplasm, with areas of clear cells. | |
| Figure 2. High-power view of the same neoplasm revealing clear cells without pronounced atypia and pigment production. | |
|
|
| Figure 3 | Figure 4 |
|---|---|
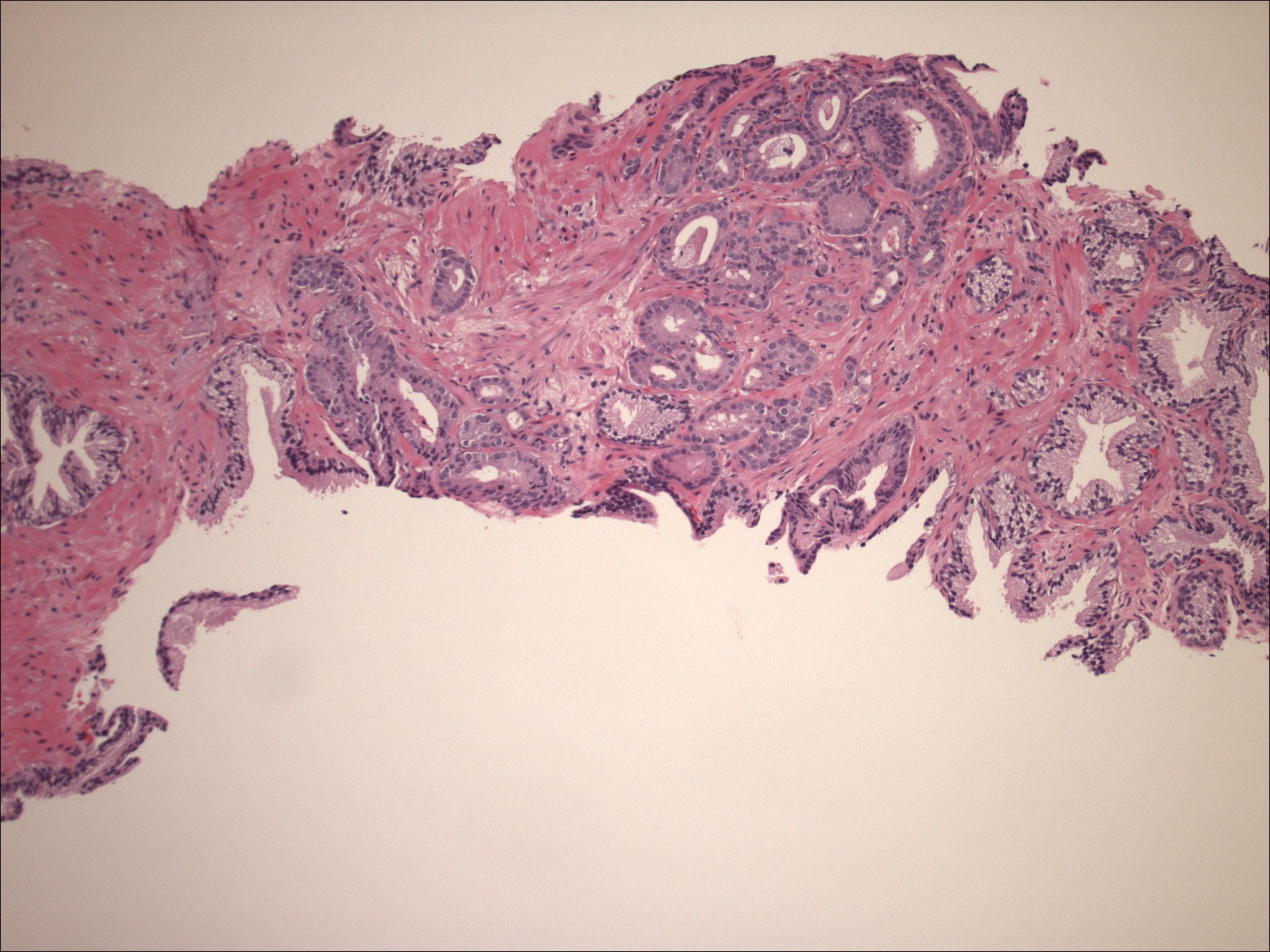
| Figure 3. Needle biopsy of the prostate, performed in 1999, revealing prostatic adenocarcinoma, acinar type, Gleason grade 3 + 4. | |
| Figure 4. High-power view of the prostatectomy specimen, revealing the acinar adenocarcinoma of high grade (3 + 4 = 7), without any spindling or clear cell features. | |
|
|
| Figure 5 | Figure 6 |
|---|---|
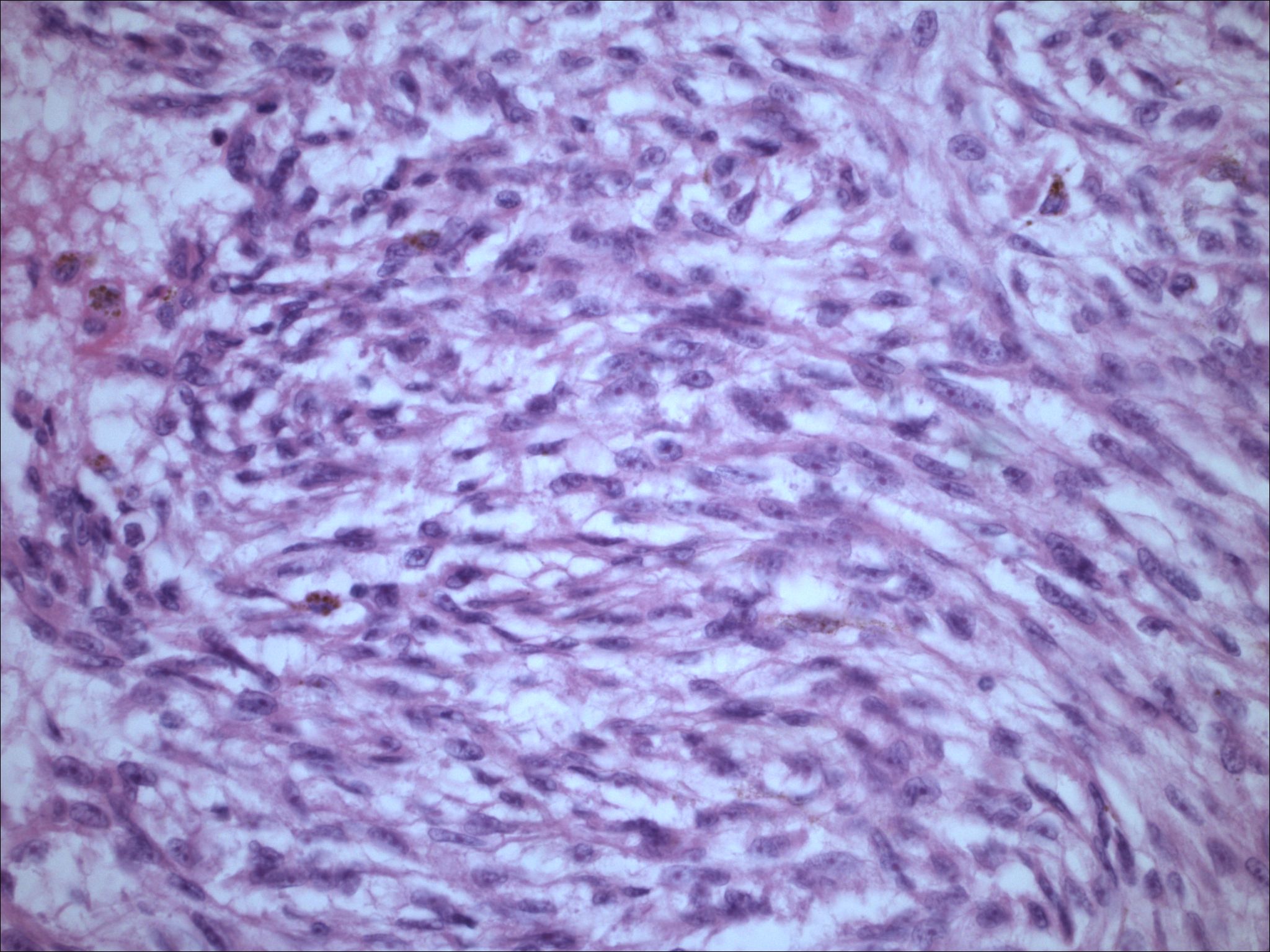
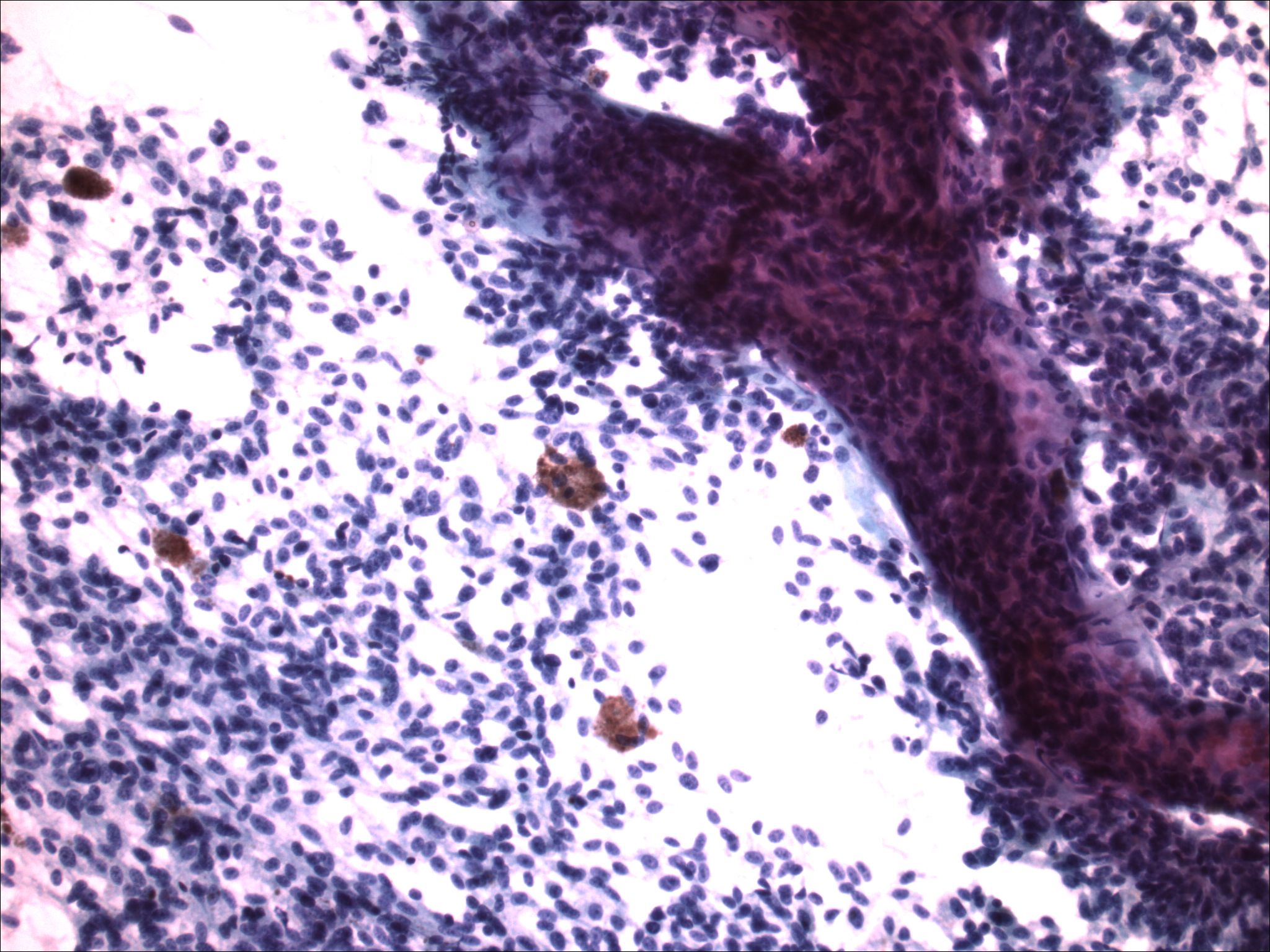
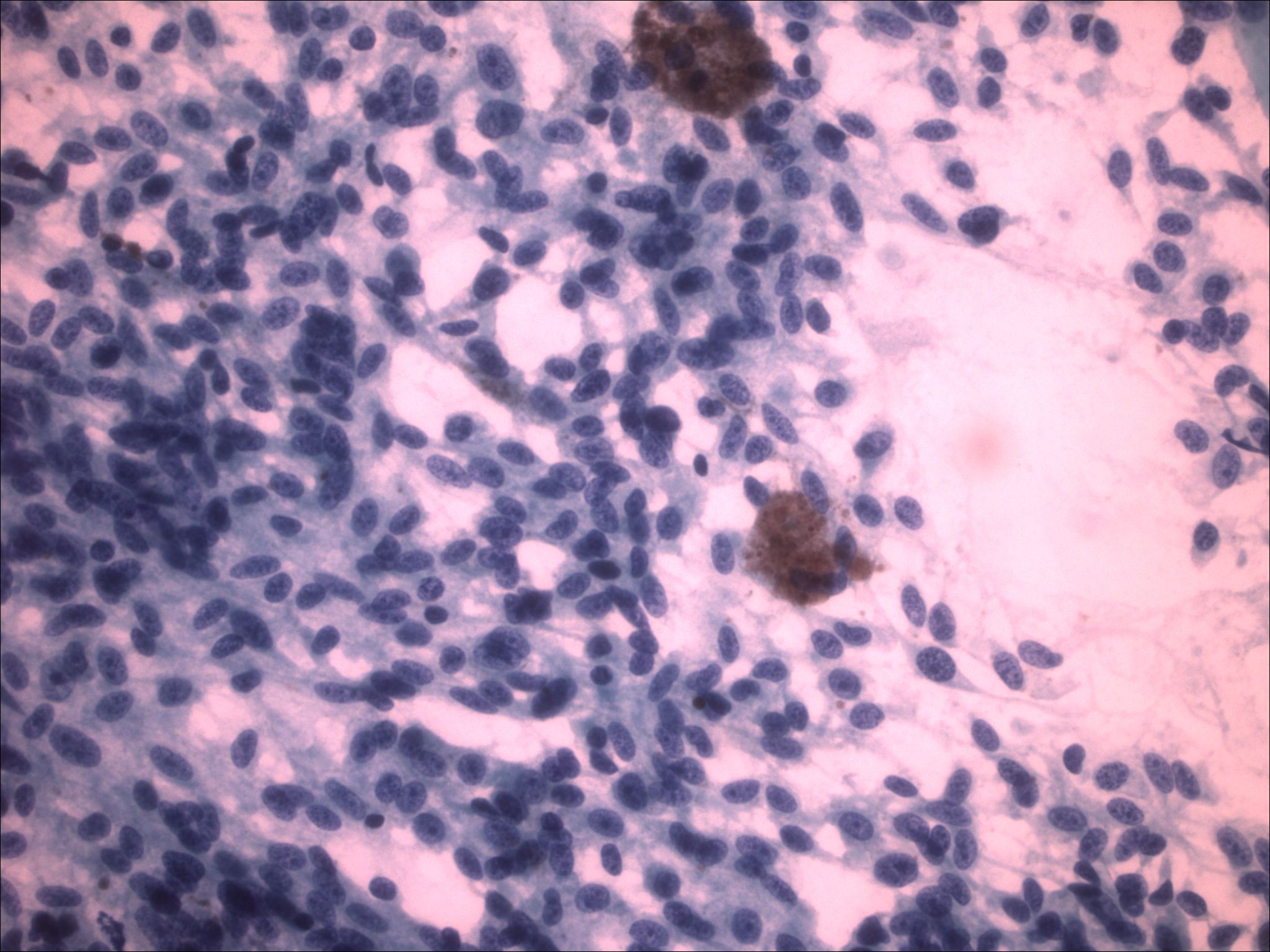
| Figure 5. Medium power of the FNA specimen from the pleural cavity, revealing bland cells with whorls of spindle shaped cells and dispersed melanin. | |
| Figure 6. High-power of the FNA specimen revealing cellular material without significant atypia, spindling, and melanin production. | |
|
|
| Figure 7 | Figure 8 |
|---|---|
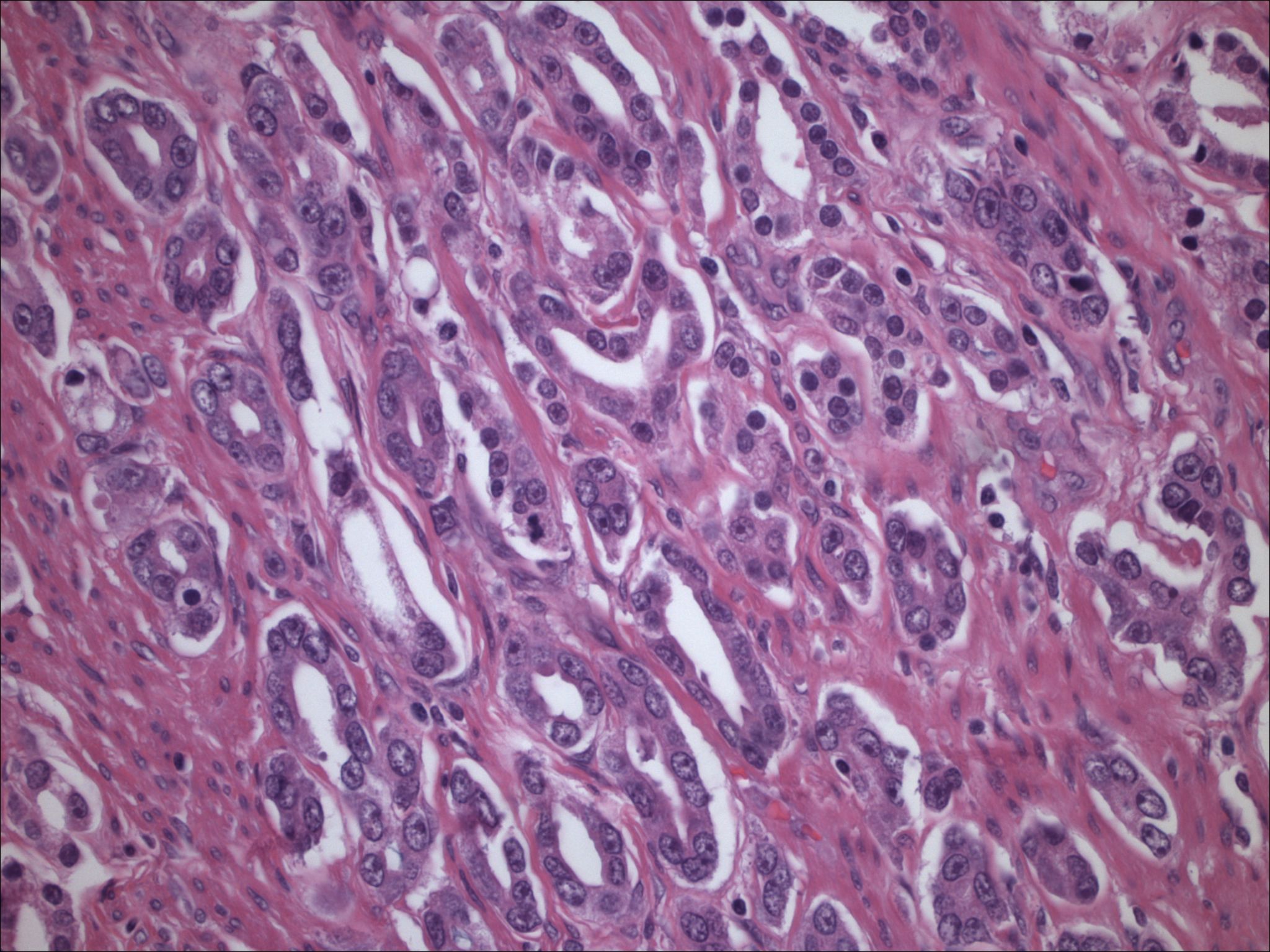
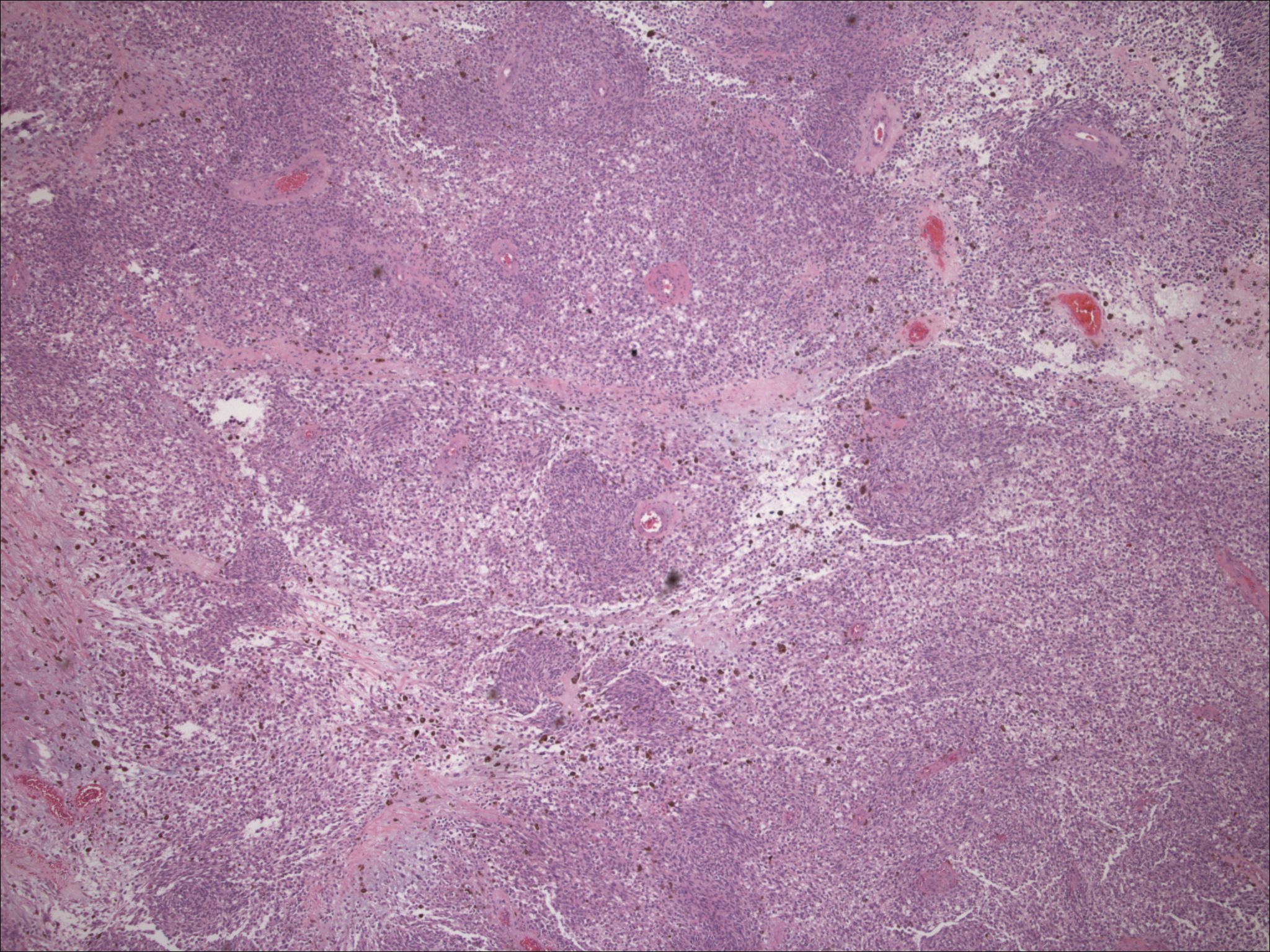
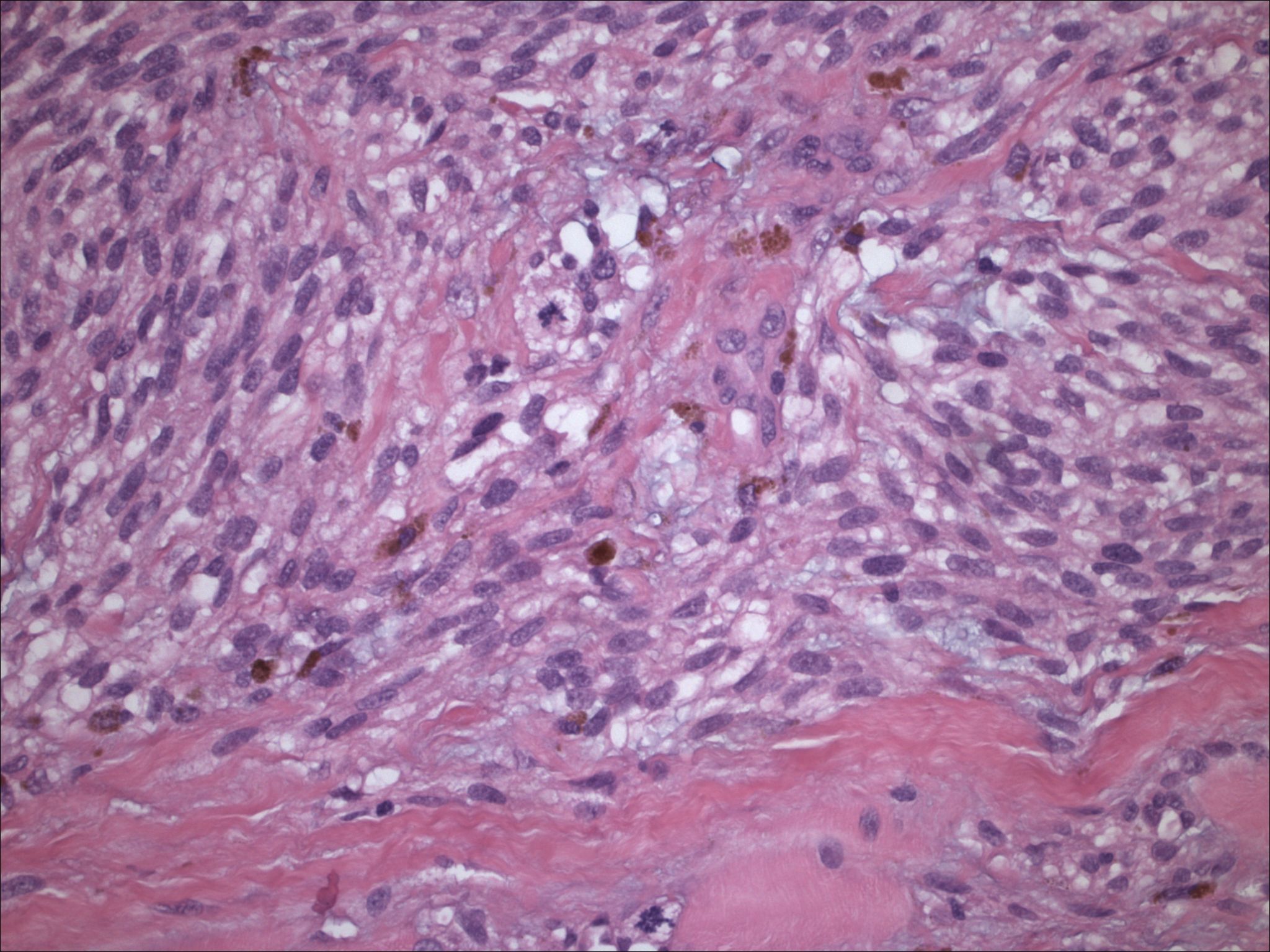
| Figure 7. Low-power view of a melanin producing neoplasm resected in 2001; material is highly cellular with areas of clearing. | |
| Figure 8. Medium power of spindle-shaped and clear cells, with focal melanin production, low mitotic activity, and increase in vasculature. | |
|
|
| Figure 9 | Figure 10 |
|---|---|
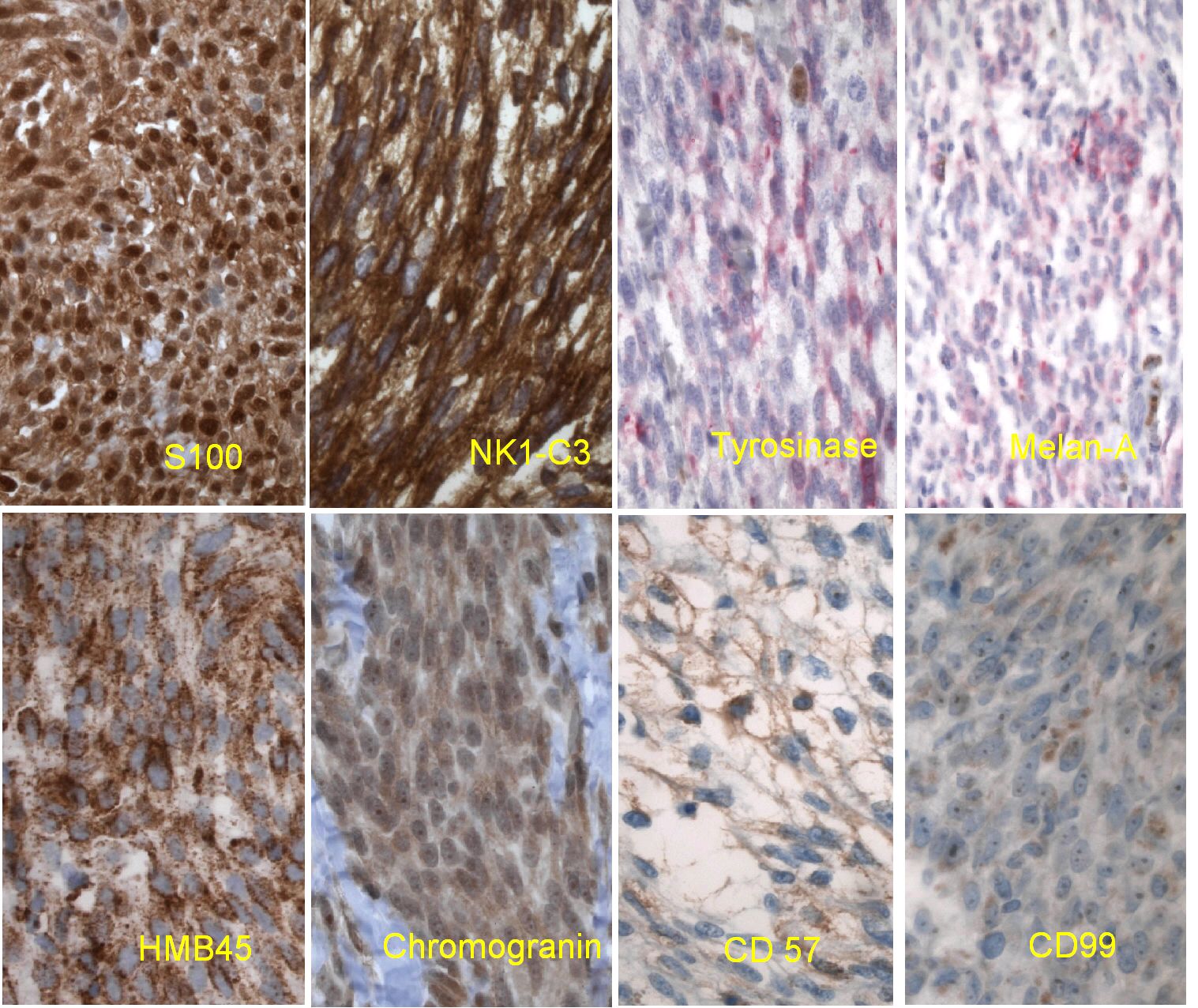
| Figure 9. Composite view of the positive immunohistochemical stains in this specimen, combining the brown chromogen and red chromogen positivity. | |
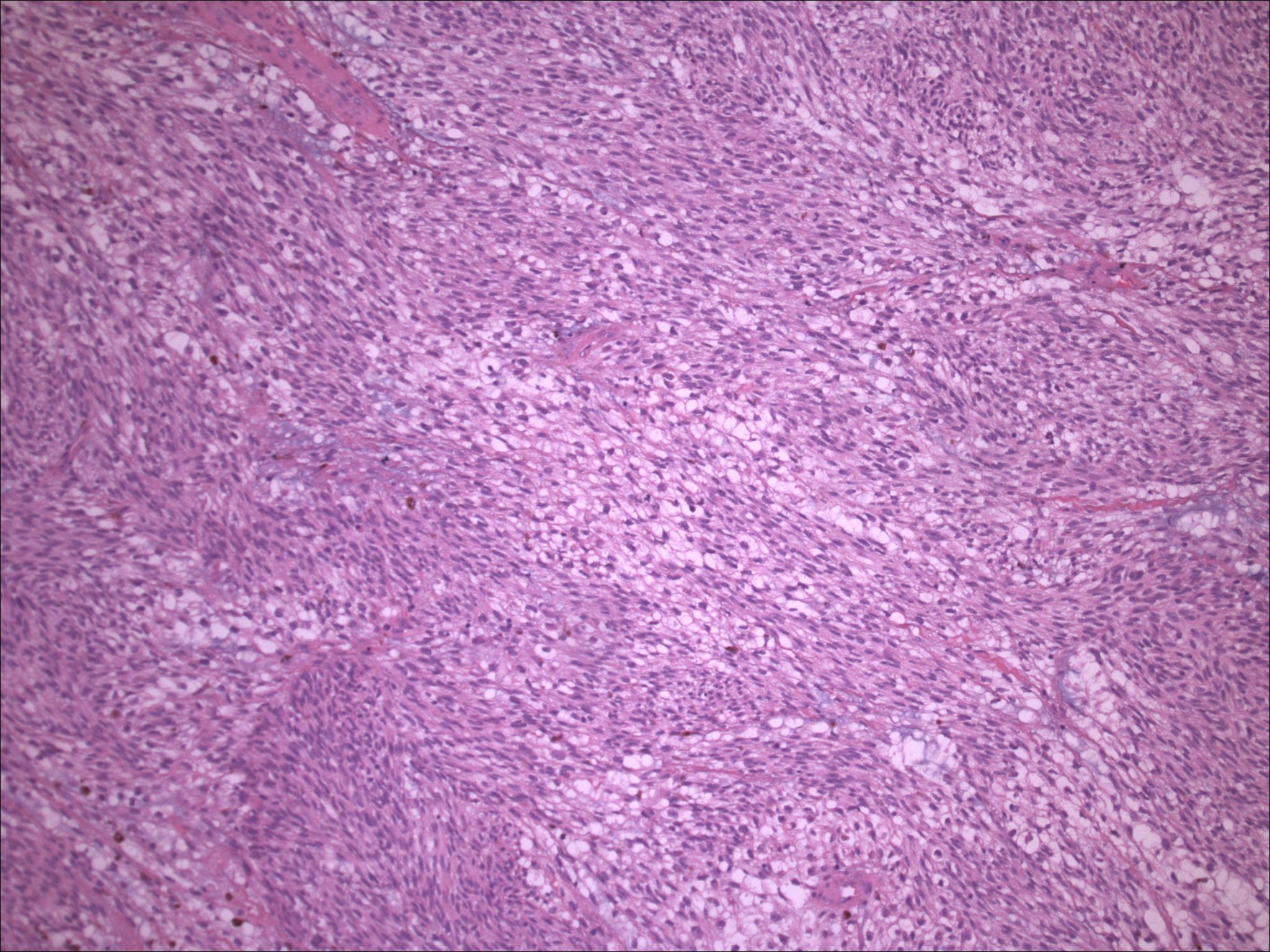
| Figure 10. Clear cell sarcoma, metastatic, high power, with rare mitotic figures, melanin production, spindle and clear cells and no nucleoli. | |
Initial histopathologic impression, on the tissue obtained in 1988 was that of a high-grade spindle cell neoplasm infiltrating or possibly replacing the lymph node (Figs. 1-3); the diagnosis (with the aid of electron microscopy) was spindle cell carcinoma with neuroendocrine differentiation. Malignant melanoma was considered to be an option but there was no history of a primary cutaneous malignancy. After subsequent development of a pulmonary nodule in 2001, and fine needle aspirate (FNA), H&E stain performed on biopsied lung and pleural tissue revealed whorls of spindle-shaped cells with blunt nuclei and clear cytoplasm, rare mitotic figures, and very slight cellular polymorphism (Figs. 5 and 6). There was focal extra- and intra-cellular pigment deposition. The neoplastic cells displayed positivity for chromogranin, synaptophysin (non-uniform), CD57 (Leu-7), S100 (strong), Melan-A (partial), Tyrosinase (partial), NK1C3 (uniform), vimentin (uniform), HMB-45 (partial), and CD99 /MIC-2/ (partial) (Fig. 9). They were negative for CD21, CD68, SMA (smooth muscle actin), CD34, Collagen IV, Laminin, Cathepsin B, GFAP, EMA, Neurofilament and CD56 (NCAM).
The histopathologic and immunohistochemical profile of this apparently melanocytic neoplasm is diagnostic of clear cell sarcoma of soft parts (CCS). Whether the neoplasm (in the original specimen) infiltrated or arose within an axillary lymph node as opposed to its development as a distinct neoplasm in the axillary region is unclear. However, we could not demonstrate an association with a nerve (perineural location), and there was no identifiable primary cutaneous neoplasm that would, perhaps, give rise to the metastatic deposit.
We also examined his prostate pathology (both biopsy and prostatectomy) (Figs. 3 and 4), which revealed poorly differentiated prostatic adenocarcinoma of acinar type, Gleason grades 3 + 4, score 7 with no apparent neuroendocrine or spindle cell differentiation, ruling out this malignancy as a possible primary.
Differential diagnosis
The differential diagnosis of a neoplasm such as the one presented here, include CCS, MM, follicular dendritic and interdigitating cell tumors (IDCT/FDCT), malignant peripheral nerve sheath tumor (MPNST), and a category of so-called Pecomas; these should be entertained, based on their similarities in terms of embryologic origin and histology, and expression of immunohistochemical markers. However, subtle but distinguishing differences have been reported in the literature, potentially allowing one to make a final diagnosis.
Discussion:
Clear cell sarcoma
The entity of CCS is also known as malignant melanoma of soft parts [1]. According to some authorities, the proper name for this particular neoplasm should be clear cell sarcoma of tendons and aponeuroses, in order to prevent confusion with clear cell sarcoma of the kidney and other clear and spindle cell neoplasms.
Perineural cells, Schwann (neurilemmal) cells, and melanocytes all share a common embryologic origin: that of neural crest cells. Owing to their common origin, a number of markers, importantly S100, are expressed by both cell and tumor types. Pigmented neuroendocrine tumors with focal expression of both S100 and HMB-45 have been described, and stains for synaptophysin and chromogranin have been incorporated as potential distinguishing markers.
In recent years, the characteristic translocation t (12; 22)(q13; q13), which results in a fusion gene EWS/ATF1 2, has been deemed pathognomonic for these neoplasms. To date, this translocation has not been identified in the cutaneous malignant melanomas (MMs), or malignant peripheral nerve sheath tumors (MPNSTs), which are also derived from neural crest. Therefore, there was a push to reclassify (or keep classifying) this neoplasm as a separate entity, apart from the melanoma group, mainly based on this unique translocation. The EWS-ATF1 fusion gene encodes a constitutive transcriptional activator [3 4, 5].
Other histopathologic criteria that support the classification of CCS as a separate entity include the presence of spindle and clear cells, absence of nuclear atypia, and small and inconspicuous nucleoli. Clinically, CCS most often presents in young adults, with a slight female predominance, as a slow-growing but painful nodule on the extremities, very commonly on the ankle or foot. Primary CCS usually arises in the deeper soft tissues, in association with fascia, tendons, or aponeuroses, sometimes with extension into subcutis or dermis. In contrast, it is rather unusual (although not unheard of) for malignant melanoma to present without an identifiable primary skin lesion, and once metastatic foci have been identified in one anatomical location, other sites of concurrent metastasis would almost be expected. In cases of metastatic melanoma in which a cutaneous neoplasm is not identified upon initial presentation, historical evidence of a regressed primary skin lesion should be sought. CCS has a propensity to spread over the course of a number of years to regional lymph nodes, lung, or bone. The 5-year survival rates have been estimated to range of 48-67 percent, but prognosis can be dismal if widely metastatic at diagnosis [6]. Histologically, MM tends to be more mitotically active, displaying significant nuclear pleomorphism and cellular atypia, than CCS. It has been suggested that CCS can be conclusively diagnosed by collectively using cytology, immunohistochemistry (HMB-45+ S-100+), cytogenetic analysis (demonstrating the specific translocation) and EM to supply ultrastructural evidence for the presence of melanosomes. However, CCS shares the same immunohistochemical profile with MM, with two exceptions: (1) CCS is positive for synaptophysin (while melanoma is negative) [7], and (2) CCS displays occasional positivity for chromogranin while MM does not (Table 1).
Malignant peripheral nerve sheath tumors
MPNST are usually found in association with nerve trunks, and to find MPNST originating in or metastasizing to lymph nodes is exceedingly rare. MPNST characteristically have a basal lamina around tumor cells (which usually reveals positivity with laminin and collagen IV), and is distinguishable by electron microscopy. Additionally, when MPNST are found to express S100, the expression is usually faint and focal. In contrast, melanomas tend to express S100 diffusely, in both the nuclear and cytoplasmic compartments. Histologically, schwannomas and MPNSTs can have spindle shaped nuclei, rendering them difficult to distinguish from spindle-type melanomas, especially when these perineural cell tumors display numerous mitoses.
| Marker | Malignant Melanoma |
Clear Cell Sarcoma |
Malignant peripheral nerve sheath tumor |
| S100 | +++ | +++ | ++ |
| HMB45 | ++ | ++ | - * |
| Melan-A/MART1 | +++ | ++ | - * |
| Tyrosinase | +++ | ++ | - * |
| Synaptophysin | -/+ | + | - |
| Chromogranin | - | -/+ | - |
| NSE | ++ | +++ | ++ |
| CD56 (NCAM) | + | ++ | ++ |
| MITF-1 | ++ | + | - * |
| CD68 | +/- | - | - |
| Vimentin | +++ | +++ | +++ |
| EMA | -/+ | - | +/- |
| GFAP | + | - | + |
This group includes clear cell myoepithelial tumors of the pancreas and uterus, angiomyolipomas (both renal and extrarenal), and lymphangioleiomyomas. Histopathologically, these neoplasms are recognized by their location (usually in close proximity to vascular structures), and their cellular morphology, with epithelioid cells containing clear but faintly eosinophilic cytoplasm. Additionally, most Pecomas have been associated with the tuberous sclerosis complex (TSC), with the exception of pancreatic and uterine Pecomas. Reports of neoplasms classified generally as Pecomas or, as one of the Pecoma family members, clear cell myelomelanocytic tumors, have shown reactivity against HMB-45, smooth muscle actin, myosin, and MiTF (micro-ophthalmia inhibition transcription factor). Stage II melanosomes, with very small amounts melanin, suggestive of the presence of stage III melanosomes, have also been noted in these neoplasms, which account for their HMB-45 positivity. Importantly, these neoplasms do not co-express S-100 protein, suggesting a distinction from purely melanocytic tumors such as malignant melanomas or clear cell sarcomas. As noted above, clear cell sarcomas differ from Pecomas immunohistologically by their expression of HMB-45, MiTF, and S100 without expression of muscle markers, as well as ultrastructurally by their production of stage II and III melanosomes.
PEC (perivascular epithelioid cell)-omas:
Another entity that must be included in the differential diagnosis is a group of so-called PEComas, which are characterized by their expression of subset of melanocytic and muscle markers [8, 9],see Table 2. This group includes clear cell myoepithelial tumors of the pancreas and uterus, angiomyolipomas (both renal and extrarenal), and lymphangioleiomyomas. Histopathologically, these neoplasms are recognized by their location (usually in close proximity to vascular structures), and by cellular morphology, with epithelioid cells containing clear but faintly eosinophilic cytoplasm. Additionally, most PEComas have been associated with the tuberous sclerosis complex (TSC), with the exception of pancreatic and uterine PEComas. Reports of neoplasms classified generally as PEComas or, as one of the PEComa family members, clear cell myelomelanocytic tumors have shown reactivity against HMB-45, smooth muscle actin, myosin, and MiTF (micro-ophthalmia inhibition transcription factor). Stage II melanosomes, with very small amounts melanin, suggestive of the presence of stage III melanosomes, have also been noted in these neoplasms, which account for their HMB-45 positivity. Importantly, these neoplasms do not co-express S-100 protein, suggesting a distinction from purely melanocytic tumors such as malignant melanomas or clear cell sarcomas. As noted above, clear cell sarcomas differ from PEComas immunohistologically by their expression of HMB-45, MiTF, and S100 without expression of muscle markers, as well as ultrastructurally by their production of stage II and III melanosomes.
Follicular dendritic cell and interdigitating cell tumors
Histologically, and due to the possible location in the lymph node, we have also considered the differential diagnoses of follicular dendritic cell (FDC) and interdigitating cell (ITDC) tumors, especially since these neoplasms (more so with ITDC tumors) are often positive for S-100. These neoplasms are phenotypically similar to antigen presenting cells. Follicular dendritic cell tumors typically express CD21, CD35, Ki-M4P, and Ki-FDRC1p (Table 2). Histologically, they can be recognized as fascicles and whorls of oval-to-spindle shaped cells with minimal mitotic activity. There is usually a population of lymphocytes distributed among the tumor cells. As outlined above, these neoplasms can be positive for S100, although expression of this marker is not consistent. Most of the previously reported cases of interdigitating dendritic cell tumors have arisen in the lymph nodes. Although ITDC tumors can appear histologically similar to follicular DCs, immunohistochemical stains allow distinction between these tumor types. Interdigitating DC tumor cells express S100 and CD68 (macrophage marker), but are negative for CD21 and CD35 (Table 2).
| Marker | Follicular Dendritic Tumor |
Interdigitating Cell Tumor |
PEComas |
| S100 | +/- | ++ | +++ |
| CD21 | ++ | - | - |
| CD35 | ++ | - | - |
| CD68 | - | ++ | + |
| AMA/HHF35 | - | - | ++ |
| Desmin | - | - | ++ |
| SMA | - | - | ++ |
| CD117 (c-kit) | - | - | +++ |
| Melan-A | - | - | ++ |
| HMB45 | - | - | ++ |
| Tyrosinase | - | - | ++ |
Conclusions
Initial diagnosis of CCS often rests in the hands of the dermatologist and dermatopathologist. For CCS, prognosis tends to be poor when the patients are of advanced age or the tumor nodule measures greater than 5 cm, emphasizing the importance of early recognition of potential lesions. With large tumor nodules, there is an increased risk of metastasis to local lymph nodes or the lungs. It has been well established that surgery is a critical first approach to treatment of CCS. Various chemotherapeutic regimens have been used as adjuvant therapy for recurrent or metastatic tumors, although which combination of agents to use has not been determined due to the small number of cases of CCS reported.
Clinicians should be aware that several cases are reported in the literature in which the diagnosis of CCS was apparently missed because the primary neoplastic lesion appeared to the clinician as a plantar wart, foot ulcer, or callus [10, 11]. It is the dermatopathologists role to distinguish CCS from malignant melanoma and other entities. CCS and MM require different therapy. For melanoma, wide surgical excision is recommended. Beyond this, further treatment depends on primary tumor thickness, however there is no recommended standard chemotherapy. In contrast to melanoma, combination chemotherapy following surgical excision holds promise for treatment of CCS.
Importantly, clinicians must be vigilant in their physical examinations and follow-up of patients diagnosed with these melanocytic lesions, paying particular attention to local recurrences or lymph node metastasis, both of which impact on overall survival rates, and response to treatment.
As a final word, it is important to be aware of the number of differential diagnoses associated with clear- and spindle-cell neoplasms; only by carefully considering and excluding each entity should one give the diagnosis of "metastatic melanoma of unknown primary". This diagnosis should not be a waste-basket for incompletely worked up tumors; the multitude of immunohistochemical markers available should be utilized.
References
1. Chung EB, Enzinger FM. Malignant melanoma of soft parts. A reassessment of clear cell sarcoma. Am J Surg Pathol 1983; 7:405-13.2. Antonescu CR, Tschernyavsky SJ, Woodruff JM, Jungbluth AA, Brennan MF, Ladanyi M. Molecular diagnosis of clear cell sarcoma: detection of EWS-ATF1 and MITF-M transcripts and histopathological and ultrastructural analysis of 12 cases. J Mol Diagn 2002; 4:44-52.
3. Brown AD, Lopez-Terrada D, Denny C, Lee KA. Promoters containing ATF-binding sites are de-regulated in cells that express the EWS/ATF1 oncogene. Oncogene 1995; 10:1749-56.
4. Fujimura Y, Ohno T, Siddique H, Lee L, Rao VN, Reddy ES. The EWS-ATF-1 gene involved in malignant melanoma of soft parts with t(12;22) chromosome translocation, encodes a constitutive transcriptional activator. Oncogene 1996; 12:159-67.
5. Fujimura Y, Siddique H, Lee L, Rao VN, Reddy ES. EWS-ATF-1 chimeric protein in soft tissue clear cell sarcoma associates with CREB-binding protein and interferes with p53-mediated trans-activation function. Oncogene 2001; 20:6653-9.
6. Lucas DR, Nascimento AG, Sim FH. Clear cell sarcoma of soft tissues. Mayo Clinic experience with 35 cases. Am J Surg Pathol 1992; 16:1197-204.
7. Swanson PE, Wick MR. Clear cell sarcoma. An immunohistochemical analysis of six cases and comparison with other epithelioid neoplasms of soft tissue. Arch Pathol Lab Med 1989; 113:55-60.
8. Bonetti F, Pea M, Martignoni G, Zamboni G. PEC and sugar. Am J Surg Pathol 1992; 16:307-8.
9. Pea M, Bonetti F, Zamboni G, et al. Melanocyte-marker-HMB-45 is regularly expressed in angiomyolipoma of the kidney. Pathology 1991; 23:185-8.
10. Gill J, el-Azhary R, Davis M. An atypical, painful foot ulcer. The International Society of Dermatology 2002; 41:692-693.
11. Fujimoto M, Hiraga M, Kiyosawa T, et al. Complete remission of metastatic clear cell sarcoma with DAV chemotherapy. Clinical and Experimental Dermatology 2002; 28:22-24.
© 2004 Dermatology Online Journal